Arabidopsis thaliana
Taxonomy:Eukaryota; Viridiplantae; Streptophyta; Streptophytina; Embryophyta; Tracheophyta; Euphyllophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicotyledons; rosids; eurosids II; Brassicales; Brassicaceae; Arabidopsis
Introduction
For over two decades, Arabidopsis thaliana is the model system of choice for plant molecular geneticists (Meinke et al., 1998). Although it has been shown that it has undergone several ancient polyploidy events in its evolutionary past (Vision et al., 2000; Simillion et al., 2002; Blanc et al., 2002; Bowers et al., 2003), the genome of Arabidopsis remained small, i.e. about 1/25th of the human genome, which made it a good candidate for sequencing the first plant genome. Since the completion of the genome sequence project four years ago (The Arabidopsis Genome Initiative, 2000), and many updates of the annotation later, biologists and bioinformaticians still devote much of their time to improve and decode the content of the five Arabidopsis chromosomes. The correct description of the genes and their structure is the necessary prerequisite for many functional genomics experiments, including reverse genetics, transcript profiling and proteomics approaches, as well as any comparative analysis with other genomes. Much progress is being made, and because neither intrinsic nor extrinsic approaches are able to produce the exhaustive proteome for a single genome, newer programs have been developed, focusing on combining both types of approaches. To automatically generate a new full genome annotation for Arabidopsis that combines intrinsic gene prediction with the latest experimental data, we used the gene prediction platform EuGène, a flexible generic gene finding system for eukaryotic genomes. The novelty of this tool is that it integrates both extrinsic data and intrinsic predictions from the very beginning of the process, exploiting the available sequence homology to direct gene structure prediction. This approach enables the integration of different independent 'ab initio' modules, each performing at their best for particular steps during the prediction, and lets the EuGène software decide based on weighted scores which path to follow, taking protein homologies and spliced alignments with EST and cDNA sequences into account. As such, this software tool also minimizes the 'a posteriori' manual or automated integration and curation of extrinsic data.
Downloads
Materials and MethodsThe EuGene genome annotation platform
EuGène is a flexible generic gene finding system for eukaryotic organisms that combines extrinsic sequence information contained in cDNA and protein databases with intrinsic information obtained from signal and content sensor systems. The strength of EuGène lies in the fact that it integrates all these sources of information in one gene model represented by a directed acyclic weighted graph (DAG; see Fig. 1). This DAG represents all possible consistent gene structures by a path. By assigning weights to each of the edges in the DAG, a shortest path in this graph can be found that is associated with the optimal gene-structure. 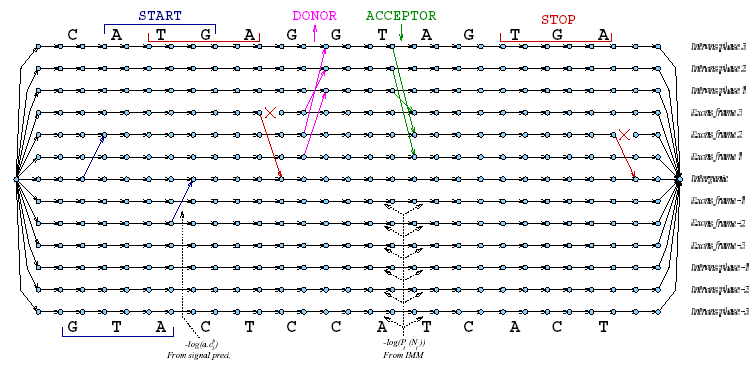
Figure 1: The DAG gene model explored by EuGene for a simple sequence
The signal sensors (or graph-edges) used were SplicePredictor and NetGene2 for splice site prediction and the NetStart for ATG prediction.
For the content sensor that predicts coding sequences, five interpolated Markov models are trained: one for generating sequences in each of the three reading frames, one for generating introns and one for generating intergenic sequences. These IMMs allow to compute the probability Pt(Ni) that the nucleotide Ni at position i appears on each track t. The corresponding edge is weighted -log (Pt(Ni)) as shown in Figure 1. The contribution of each of these systems in the gene model is globally optimized to maximize gene prediction accuracy on a set of annotated sequences. Extrinsic information from cDNA and protein databases is used in the DAG by prohibiting certain paths to be followed. cDNA alignments in conjunction with splice site information are used to modify the graph by forbidding paths that would be incompatible with the cDNA data. Protein homology information is used in a similar way. However, one cannot be confident enough in such information to directly use matches/gaps as constraints and we therefore simply modify the Pt (Ni) using a simple pseudo-count scheme. EuGene also takes into account the existence of repeated regions as found by the RepeatMasker system as well as the existence of frameshifts.
Given an adequate graph, a simple linear time, linear space shortest path algorithm such as Bellman’s algorithm can output the best possible gene structure. We use a slightly more sophisticated algorithm that can take into account constraints on the minimum length of some gene elements (introns, single exon genes, intergenic regions). In practice - EuGene: An Eucaryotic Gene Finder that combines several sources of evidence. T. Schiex, A. Moisan and P. Rouzé. Computational Biology, Eds. O. Gascuel and M-F. Sagot, LNCS 2066, pp. 111-125, 2001. Link: www.inra.fr/mia/T/EuGene
- NetStart (version 1.0, option –at ; Pedersen and Nielsen, 1997)
- NetGene2 (version 2.42, options -a –e –p –r –s at ; Tolstrup et al. 1997)
- Splice Predictor (version 25/06/97, options -f –c 1 –m 1 –l 0 ; Brendel and Kleffe, 1998)
- blastx (NCBI version 2.2.2, options –e 1e-6 –b 500000 –v 500000 –G 32767 –E 32767 ; we did not use the ungapped blast because we noticed it was bugged)
- sim4 (version 03/03/2002, options A=6 P=1).
- RepeatMasker version 071302, Repbase version 7.8, with option –nolow. Additionally, hits identified by RepeatMasker, smaller than 50 nucleotides were not taken into consideration.
Extrinsic data - Swissprot release 40
- PIR release 74_03
- EST and Full length cDNAs (all transcripts available 20/06/2003)
Results
The annotation of the Arabidopsis genome provided here is one performed using the EuGène platform (v 1.5.8) on the pseudo molecules from TiGR version 3 released in 2002. The reason for using this older version was mainly because of development and evaluations purposes. Nevertheless, we compared our results with the latest available TiGR annotation performed on the 4 th version of their genome assembly. This means that for a certain small percentage of the used pseudo-molecules we missed the genomic sequences that were integrated into the latest assembly. This represents an overall small quantity that should not bias our comparison dramatically. Also rearrangements of BACs due to novel sequences along the pseudo-molecules might affect gene predictions locally, but would stay globally nominal. The amount and diversity of ESTs and cDNAs used differ also, as here too, we kept our extrinsic data set as stable as possible. This means that for different genes we might have a prediction solely based in intrinsic sensors while meanwhile a cDNA or EST became available. In order to be able to compare with up to date TiGR annotation we extracted all the genes predicted by EuGène, post processed them to add the missing overlapping UTRs, as those needed to be removed to avoid that EuGène would fuse different genes, and mapped them again on the pseudo-molecules version 4 from TiGR. The EuGène genes mapped on the TiGR v4 pseudo-molecules were then compared with the gene structures as they are annotated on those sequences using the FLAGdb++ database. The comparison reported 22394 (82.4%) of the genes that were seen by both annotations, and from which 12921 (57.7%) of the genes that were, from start to stop of the CDS, completely identical in every respect. The main concentrations of divergent gene structures were, as was expected in the heterochromatine, around the centromeres, while in the euchromatine minimal differences were observed (see fig). 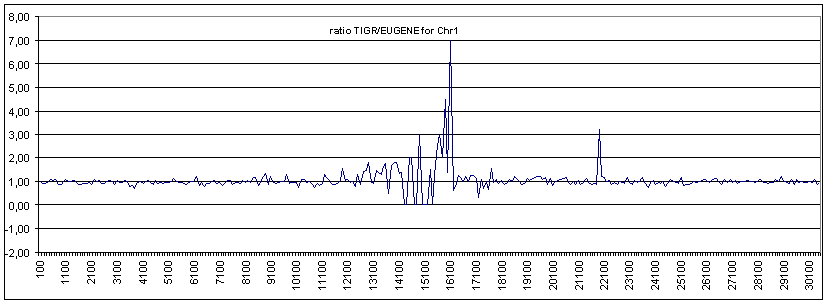
The number of genes that were predicted by TiGR and not seen by EuGène were counted up to 3311, but only a fraction (81 genes) of those was as expected due to genomic sequence not being available at the time the EuGène prediction was done. On the reverse EuGène predicted 1525 genes that were not seen at all by TiGR. The 3230 genes found by TiGR and not seen by EuGène were checked for the presence of a known protein domain using the PFAM library. This resulted in 1504 genes with a protein domain, or 45% of this subset of genes, which is compared to the total number of genes predicted by TiGR having a known domain lower than the mean value of 67%. Surprisingly, from the 1525 genes only predicted by EuGène 700 (46%) appeared to have a cognate EST or cDNA. For the reverse situation, 2686 out of the 3311, or 81% of the genes only predicted by TiGR did have a cognate transcript. The latter is likely due to the lacking data at the time of the prediction done using EuGène. The remaining genes were either for a minor fraction on opposite strands or were split in two or more genes while the counter part predicted a single gene at the same location (see table). A comparison at mRNA level has also been done, but reported only 5995 (22%) of genes completely identical from transcription start to end of the 3’UTR. This discrepancy between mRNA-level and CDS-level mainly reflects the differences in ESTs and cDNAs used and is thus not a good criterium for comparison. To compare both platforms independently of the availability of any transcript that would have influenced the prediction, we made the same comparison counting only genes (set of 5327 in total) that did not have any EST or cDNA aligning with them. As expected the number of genes completely identical in every respect drop to 42.7% (2275 genes). People Involved
Carine Serizet 1,2, Stephane Rombauts 1, Sven Degroeve 1, Vincent Thareau 1,2, Thomas Schiex 3, Véronique Brunaud 2, Sébastien Aubourg 2, Pierre Rouzé 1, and Yves Van de Peer 1 1Department of Plant Systems Biology, Flanders Interuniversity Institute for Biotechnology (VIB), Ghent University, Technologiepark 927, B 9052 Gent, Belgium 2Unité de Recherche en Génomique Végétale (INRA CNRS UEVE), 2 rue Gaston Crémieux, CP 5708, F 91057 Evry CEDEX, France 3INRA Toulouse, Mathematics and Computer sciences for life sciences. Chemin de Borde Rouge, BP 27 Castanet-Tolosan 31326 Cedex, France Acknowledgements GPI : E. Barillot, F. Legeai et S. Reboux Flagdb : V. Brunaud Gent : Eric Bonnet Update blastn : F. Samson CATMAdb : Mark Crowe The annotation work was supported by the grant No-2000-009 from the French Génoplante program to Pierre Hilson and Pierre Rouzé.
Publications
2015Szakonyi, D., Van Landeghem, S., Baerenfaller, K., Baeyens, L., Blomme, J., Casanova-Sáez, R., De Bodt, S., Esteve-Bruna, D., Fiorani, F., Gonzalez, A., Grønlund, J., G.H. Immink, R., Jover-Gil, S., Kuwabara, A., Muñoz-Nortes, T., D.J. van Dijk, A., Wilson-Sánchez, D., Buchanan-Wollaston, V., C. Angenent, G., Van de Peer, Y., Inzé, D., Luis Micol, J., Gruissem, W., Walsh, S., Hilson, P. (2015)
The KnownLeaf literature curation system captures knowledge about Arabidopsis leaf growth and development and facilitates integrated data mining. Current Plant Biology 2:1-11.
Ghorbani, S., Lin, Y.-C., Parizot, B., Fernandez, A., Fransiska Njo, M., Van de Peer, Y., Beeckman, T., Hilson, P. (2015)
Expanding the Repertoire of Secretory Peptides Controlling Root Development with Comparative Genome Analysis and Functional Assays. Journal of Experimental Botany 66(17):5257-69.
2014Zhurov, V., Navarro, M., Bruinsma, K.A., Arbona, V., Santamaria, M.E., Wybouw, M.C.N., Osborne, E.J., Ens, C., Rioja, C., Vermeirssen, V., Rubio-Somoza, I., Krishna, P., Diaz, I., Schmid, M., Gómez-Cadenas, A., Van de Peer, Y., Grbic, M., Clark, R.M., Van Leeuwen, T., Grbic, V. (2014)
Reciprocal responses in the interaction between Arabidopsis and the cell-content feeding chelicerate herbivore Tetranychus urticae. Plant Physiol. 164(1):384-399.
Morreel, K., Saeys, Y., Dima, O., Lu, F., Van de Peer, Y., Vanholme, R., Ralph, S., Vanholme, B., Boerjan, W. (2014)
Systematic Structural Characterization of Metabolites in Arabidopsis via Candidate Substrate-Product Pair Networks. The Plant Cell 26(3):929-945.
* Pajoro, A., * Biewers, S., * Dougali, E., * Leal Valentim, F., * Adelina Mendes, M., * Porri, A., Coupland, G., Van de Peer, Y., van Dijk, A.D.J., Colombo, L., Davies, B., Angenent, G.C. (2014)
The (r)evolution of gene regulatory networks controlling Arabidopsis plant reproduction, a two decades history. Journal of Experimental Botany 65(17):4731-4745. *contributed equally
Vermeirssen, V., De Clercq, I., Van Parys, T., Van Breusegem, F., Van de Peer, Y. (2014)
Arabidopsis ensemble reverse-engineered gene regulatory network discloses interconnected transcription factors in oxidative stress. The Plant Cell 26(12):4656-4679.
2013Van Landeghem, S., De Bodt, S., Drebert, Z. J., Inzé, D., Van de Peer, Y. (2013)
The potential of text mining in data integration and network biology for plant research: a case study on Arabidopsis. The Plant Cell 25(3):794-807.
Nystedt, B., Street, N. R., Zuccolo, A., Lin, Y.-C., Wetterbom, A., Vezzi, A., Scofield, D. G., Delhomme, N., Alexeyenko, A., Giacomello, S., Vicedomini, R., Sahlin, K., Sherwood, E., Elfstand, M., Gramzow, L., Holmberg, K., Hällman, J., Keech, O., Klasson, L., Koriabine, M., Kucukoglu, M., Käller, M., Luthman, J., Lysholm, F., Olson, A., Niittylä, T., Ritland, K., Rilakovic, N., Rosselló, J. A., Sena, J., Svensson, T., Talavera-López, C., Theißen, G., Vanneste, K., Tuominen, H., Zhang, J., Wu, Z., Zerbe, P., Bhalerao, R.P., Bohlmann, J., Arvestad, L., Bousquet, J., Garcia Gil, R., de Jong, P.J., Hvidsten, T. R., MacKay, J., Ritland, K., Morgante, M., Sundberg, B., Van de Peer, Y., Lee Thompson, S., Nilsson, O., Andersson, B., Lundeberg, J., Jansson, S. (2013)
The Norway spruce genome sequence and conifer genome evolution. Nature 497(7451):579-84.
De Clercq, I., Vermeirssen, V., Van Aken, O., Vandepoele, K., Murcha, M., Law, S., Inzé, A., Ng, S., Ivanova, A., Rombaut, D., Van de Cotte, B., Jaspers, P., Van de Peer, Y., Kangasjarvi, J., Whelan, J., Van Breusegem, F. (2013)
The membrane-bound NAC transcription factor ANAC013 is a regulator of mitochondrial retrograde regulation of the oxidative stress response in Arabidopsis. The Plant Cell 25(9):3472-90.
2012Fawcett, J., Rouzé, P., Van de Peer, Y. (2012)
Higher intron loss rate in Arabidopsis thaliana than A. lyrata is consistent with stronger selection for a smaller genome. Mol. Biol. Evol. 29(2):849-59.
2011Movahedi, S., Van de Peer, Y., Vandepoele, K. (2011)
Comparative network analysis reveals that tissue specificity and gene function are important factors influencing the mode of expression evolution in Arabidopsis and rice. Plant Physiol. 156(3):1316-30.
* Fostier, J., * Proost, S., Dhoedt, B., Saeys, Y., Demeester, P., Van de Peer, Y., Vandepoele, K. (2011)
A Greedy, Graph-Based Algorithm for the Alignment of Multiple Homologous Gene Lists. Bioinformatics 27(6):749-56. *contributed equally
Proost, S., Pattyn, P., Gerats, T., Van de Peer, Y. (2011)
Journey through the past: 150 million years of plant genome evolution. Plant J. 66(1):58-65.
* Hu, T.T., * Pattyn, P., Bakker, E.G., Cao, J., Cheng, Y.-Y., Clarck, R.M., Fahlgren, N., Fawcette, J., Grimwood, J., Gundlach, H., Haberer, G., Hollister, J.D., Ossowski, S., Ottilar, R.P., Salamov, A., Schneeberger, K., Spannagl, M., Wang, X., Yang, L., Nasrallah, M.E., Bergelson, J., Carrington, J.C., Gaut, B.S., Schmutz, J., Mayer, K., Van de Peer, Y., Grigoriev, I., Nordborg, M., Weigel, D., Guo, Y-L. (2011)
The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43(5):476-81. *contributed equally
2010Rehrauer, H., Aquino, C., Gruissem, W., Henz, SR., Hilson, P., Laubinger, S., Naouar, N., Patrignani, A., Rombauts, S., Shu, C.-L., Van de Peer, Y., Vuylsteke, M., Weigel, D., Zeller, G., Hennig, L. (2010)
AGRONOMICS1: a new resource for Arabidopsis transcriptome profiling. Plant Physiol. 152(2):487-99.
2009De Bodt, S., Proost, S., Vandepoele, K., Rouzé, P., Van de Peer, Y. (2009)
Predicting protein-protein interactions in Arabidopsis thaliana through integration of orthology, gene ontology and co-expression. BMC Genomics 10:288.
2008Vandenbroucke, K., Robbens, S., Vandepoele, K., Inzé, D., Van de Peer, Y., Van Breusegem, F. (2008)
Hydrogen Peroxide-Induced Gene Expression across Kingdoms: A Comparative Analysis. Mol. Biol. Evol. 25(3):507-16.
Van Bel, M., Saeys, Y., Van de Peer, Y. (2008)
FunSiP : A Modular and Extensible Classifier for the Prediction of Functional Sites in DNA. Bioinformatics 24(13):1532-3.
Amoutzias, G., Van de Peer, Y. (2008)
Together We Stand: Genes Cluster to Coordinate Regulation. Dev. Cell 14(5):640-2.
2006Casneuf, T., De Bodt, S., Raes, J., Maere, S., Van de Peer, Y. (2006)
Nonrandom divergence of gene expression following gene and genome duplications in the flowering plant Arabidopsis thaliana. Genome Biol. 7(2):R13.
Vandepoele, K., Casneuf, T., Van de Peer, Y. (2006)
Identification of novel regulatory modules in dicot plants using expression data and comparative genomics. Genome Biol. 7(11):R103.
2005Degroeve, S., Saeys, Y., De Baets, B., Rouzé, P., Van de Peer, Y. (2005)
Predicting splice sites from high-dimensional local context representations. Bioinformatics 21(8):1332-8.
Vandepoele, K., Van de Peer, Y. (2005)
Exploring the plant transcriptome through phylogenetic profiling. Plant Physiol. 137(1):31-42.
* De Bodt, S., * Maere, S., Van de Peer, Y. (2005)
Genome duplication and the origin of angiosperms. Trends Ecol. Evol. 20(11):591-7. *contributed equally
Vandepoele, K., Vlieghe, K., Florquin, K., Hennig, L., Beemster, G.T.S., Gruissem, W., Van de Peer, Y., Inzé, D., De Veylder, L. (2005)
Genome-wide identification of potential plant E2F target genes. Plant Physiol. 139(1):316-28.
2004Bonnet, E., Wuyts, J., Rouzé, P., Van de Peer, Y. (2004)
Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important new target genes. Proc. Natl. Acad. Sci. USA 101(31):11511-6.
2003Saeys, Y., Degroeve, S., Aeyels, D., Van de Peer, Y. (2003)
Fast feature selection using a simple Estimation of Distribution Algorithm: A case study on splice site prediction. Bioinformatics 19 Suppl 2:ii179-88.
Vlieghe, K., Florquin, K., Vuylsteke, M., Rombauts, S., Van Hummelen, P., Van de Peer, Y., Inzé, D., De Veylder, L. (2003)
Microarray analysis of E2Fa-DPa-overexpressingplants uncovers a cross-talking genetic networkbetween DNA replication and nitrogen assimilation. J. Cell Sci. 116(Pt 20):4249-59.
Raes, J., Vandepoele, K., Saeys, Y., Simillion, C., Van de Peer, Y. (2003)
Investigating ancient duplication events in the Arabidopsis genome. J. Struct. Func. Genomics. 3(1-4):117-29.
Crowe, M.L., Serizet, C., Thareau, V., Aubourg, S., Rouzé, P., Hilson, P., Beynon, J., Weisbeek, P., Van Hummelen, P., Reymond, P., Paz-Ares, J., Nietfeld, W., Trick, M. (2003)
CATMA: a complete Arabidopsis GST database. Nucleic Acids Res. 31(1):156-8.
De Bodt, S., Raes, J., Florquin, K., Rombauts, S., Rouzé, P., Theissen, G., Van de Peer, Y. (2003)
Genomewide structural annotation and evolutionary analysis of the type I MADS-box genes in plants. J. Mol. Evol. 56(5):573-86.
2002Degroeve, S., De Baets, B., Van de Peer, Y., Rouzé, P. (2002)
Feature subset selection for splice site prediction. Bioinformatics 18 Suppl 2:S75-83.
Thijs, G., Marchal, K., Lescot, M., Rombauts, S., De Moor, B., Rouzé, P., Moreau, Y. (2002)
A Gibbs sampling method to detect overrepresented motifs in the upstream regions of coexpressed genes. J. Comput. Biol. 9(2):447-64.
Simillion, C., Vandepoele, K., Van Montagu, M., Zabeau, M., Van de Peer, Y. (2002)
The hidden duplication past of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 99(21):13627-32.
2001Thijs, G., Lescot, M., Rombauts, S., Marchal, K., De Moor, B., Moreau, Y., Rouzé, P. (2001)
A higher order background model improves the detection by Gibbs sampling of potential promoter regulatory elements in DNA sequences. Bioinformatics 17(12):1113-22.
2000Mathé, C., Dehais, P., Pavy, N., Rombauts, S., Van Montagu, M., Rouzé, P. (2000)
Gene prediction and gene classes in Arabidopsis thaliana. J. Biotechnol. 78(3):293-9.
1999Pavy, N., Rombauts, S., D?hais, P., Mathé, C., Ramana, D.V., Leroy, P., Rouzé, P. (1999)
Evaluation of gene prediction software using a genomic data set: application of Arabidopsis thaliana sequences. Bioinformatics 15(11):887-99.
Terryn, N., Heijnen, L., De Keyser, A., Van Asseldonck, M., De Clercq, R., Verbakel, H., Gielen, J., Zabeau, M., Villarroel, R., Jesse, T., Neyt, P., Hogers, R., Van Den Daele, H., Ardiles, W., Schueller, C., Mayer, K., Dehais, P., Rombauts, S., Van Montagu, M., Rouzé, P., De Vos, P. (1999)
Evidence for an ancient chromosomal duplication in Arabidopsis thaliana bysequencing and analyzing a 400-kb contig at the APETALA2 locus on chromosome 4. FEBS Lett. 445(2-3):237-45.
|


Internal Links
|