 Back to Bioinformatics Gent
Back to Bioinformatics GentAbstract
The abiotic stress response in plants is complex and tightly controlled by gene regulation. We present an abiotic stress gene regulatory network of 200,014 interactions for 11,938 target genes by integrating four complementary reverse-engineering solutions through average rank aggregation on an Arabidopsis thaliana microarray expression compendium. This ensemble performed the most robustly in benchmarking and greatly expands upon the availability of interactions currently reported. Besides recovering 1182 known regulatory interactions, cis-regulatory motifs and coherent functionalities of target genes corresponded with the predicted transcription factors. We provide a valuable resource of 572 abiotic stress modules of coregulated genes with functional and regulatory information, from which we deduced functional relationships for 1966 uncharacterized genes and many regulators. Using gain- and loss-of-function mutants of seven transcription factors grown under control and salt stress conditions, we experimentally validated 141 out of 271 predictions (52% precision) for 102 selected genes and mapped 148 additional transcription factor-gene regulatory interactions (49% recall). We identified an intricate core oxidative stress regulatory network where NAC13, NAC053, ERF6, WRKY6, and NAC032 transcription factors interconnect and function in detoxification. Our work shows that ensemble reverse-engineering can generate robust biological hypotheses of gene regulation in a multicellular eukaryote that can be tested by medium-throughput experimental validation.
Figure 1
We constructed an abiotic stress gene regulatory network (GRN) by taking the top 200,014 predictions of an average rank aggregation ensemble from four reverse engineering solutions (LeMoNe_qopt25R, LeMoNe_qopt50R, ClrR, TwixTrixR) on a dedicated Arabidopsis microarray expression compendium. The regulatory predictions were combined by rank aggregation into three ensembles: union, mean reciprocal rank and average rank. The top 200,014 predictions from the average rank ensemble made the abiotic stress GRN. Next, we clustered the abiotic stress GRN into 572 modules of coregulated genes based on the Jaccard similarity index of shared predicted transcription factors. We retained at most 10 TFs per module, those regulating the highest number of genes in the module (50% or more module genes) and displaying the highest average rank per module. Since each gene only ends up in one module, we recovered the most important regulators and functional environment for each gene in the abiotic stress response. By extensively benchmarking the GRN, both in silico and experimentally, we demonstrated its biological relevance.
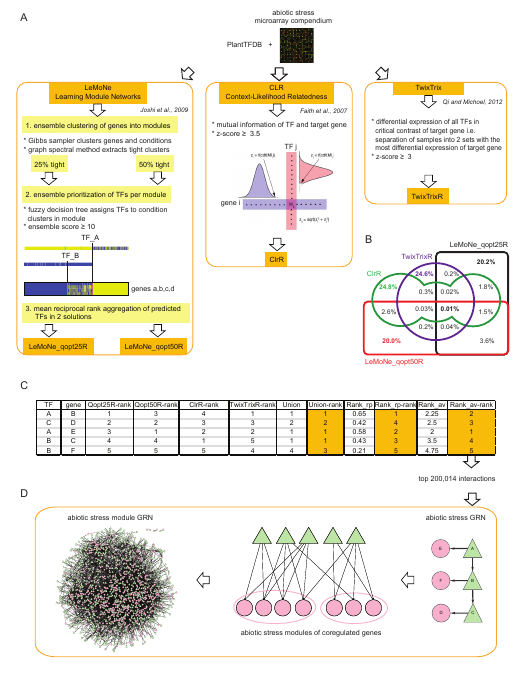
- (A) An abiotic stress microarray compendium and TFs from PlantTFDB were subjected to reverse-engineering, resulting in four network inference solutions: LeMoNe_qopt25R, LeMoNe_qopt50R, ClrR, and TwixTrixR.
- (B) The Venn diagram illustrates the percentage of 785,913 unique regulatory interactions predicted by each of the four network inference solutions and their overlap.
- (C) The regulatory predictions were combined by rank aggregation into three ensembles: union, mean reciprocal rank, and average rank.
- (D) The top 200,014 predictions from the average rank ensemble made the abiotic stress GRN. Target genes were subsequently clustered into modules of coregulated genes and only the most important regulating TFs per module (<= 10) were retained, generating the abiotic stress module GRN.