
Mission
Our objective is to extract biological knowledge from large-scale experimental data sets using data integration, comparative sequence/chromatin/expression analysis, and network biology. Through the development and application of various bioinformatics methods, we try to identify new aspects of genome biology, especially in the area of gene function prediction, gene regulation and systems biology. Whereas most of our research is focusing on plants, we are also studying gene functions and networks in green algae and diatoms. For specific examples, please see Publications.
Current research topics
Plant Gene Regulatory Networks
 Through the integration of large-scale experimental datasets and the application of computational methods, we start to have functional clues for an increasing number of genes in plants. Although new technological developments improve the resolution of measuring for example when and where a gene is expressed, understanding the underlying mechanisms and identifying the factors controlling gene expression remains a major challenge. Transcription factors (TFs) are important regulators mediating gene expression and different genomic technologies have recently been applied to start mapping the regulatory interactions between TFs and target genes in plants. As there are approximately 1700 Arabidopsis TFs and thousands of potential target genes, current gene regulatory networks remain incomplete. We develop and apply new computational approaches to identify, based on raw data from ChIP-chip/Seq, ATAC-Seq, protein binding microarrays, TF perturbation data or phylogenetic footprinting, new TF-target interactions and perform systematic regulatory gene annotations. By combining specific expression bulk and single-cell datasets with newly inferred regulatory interactions, we offer new approaches to explore condition-specific gene regulatory networks and shed light on the functions of TFs in different biological contexts. Furthermore, through sequence and expression-based comparative genomics methods, we also characterize regulatory sequences in crop species.
Through the integration of large-scale experimental datasets and the application of computational methods, we start to have functional clues for an increasing number of genes in plants. Although new technological developments improve the resolution of measuring for example when and where a gene is expressed, understanding the underlying mechanisms and identifying the factors controlling gene expression remains a major challenge. Transcription factors (TFs) are important regulators mediating gene expression and different genomic technologies have recently been applied to start mapping the regulatory interactions between TFs and target genes in plants. As there are approximately 1700 Arabidopsis TFs and thousands of potential target genes, current gene regulatory networks remain incomplete. We develop and apply new computational approaches to identify, based on raw data from ChIP-chip/Seq, ATAC-Seq, protein binding microarrays, TF perturbation data or phylogenetic footprinting, new TF-target interactions and perform systematic regulatory gene annotations. By combining specific expression bulk and single-cell datasets with newly inferred regulatory interactions, we offer new approaches to explore condition-specific gene regulatory networks and shed light on the functions of TFs in different biological contexts. Furthermore, through sequence and expression-based comparative genomics methods, we also characterize regulatory sequences in crop species.
Image: Gene regulatory network inferred through conserved noncoding sequence analysis and condition-specific co-expression information. Source: Van de Velde et al., 2014
Comparative functional genomics
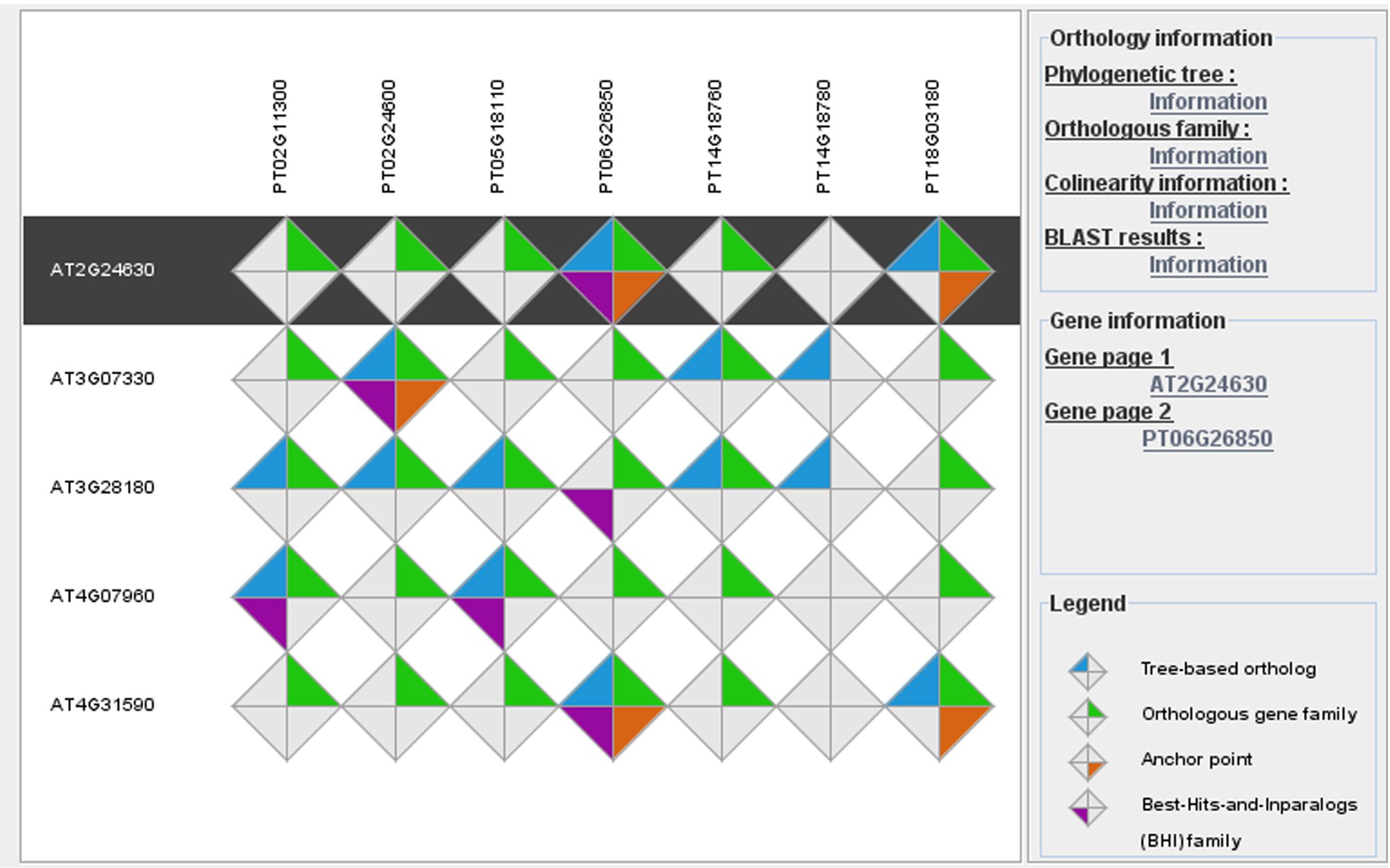 Plant genomes contain an extremely large number of genes, both in number of gene families as well as copy number per family. Consequently, searching for individual genes with conserved biological functions across different plant species is challenging. When and where a gene is expressed is an important factor contributing to the concept of gene function in multicellular organisms. Therefore, comparing expression data between different species offers a valuable alternative approach, apart from protein similarity, to identify genes showing conserved expression profiles and, as such, to translate detailed biological knowledge from model species to crops. Apart from comparative sequence analysis tools (e.g. PLAZA), we develop new methods to integrate experimental information obtained through complementary -omics approaches and to identify new genes and functional modules involved in specific biological processes.
Plant genomes contain an extremely large number of genes, both in number of gene families as well as copy number per family. Consequently, searching for individual genes with conserved biological functions across different plant species is challenging. When and where a gene is expressed is an important factor contributing to the concept of gene function in multicellular organisms. Therefore, comparing expression data between different species offers a valuable alternative approach, apart from protein similarity, to identify genes showing conserved expression profiles and, as such, to translate detailed biological knowledge from model species to crops. Apart from comparative sequence analysis tools (e.g. PLAZA), we develop new methods to integrate experimental information obtained through complementary -omics approaches and to identify new genes and functional modules involved in specific biological processes.
Image: PLAZA integrative orthology viewer. Source: PLAZA 2.5 - Van Bel et al., 2012.
Other research interests cover:
- The evolution and integration of species-specific, HGT and lincRNA genes in regulatory networks
- Integrative approaches to translate -omics data into biological knowledge
- Tool development to accurately translate biological networks between organisms with different levels of genome complexity