General documentation
Introduction
TRAPID is an online tool for the fast and efficient processing of assembled RNA-Seq (meta)transcriptome data. TRAPID offers high-throughput annotation, ORF prediction, frameshift detection and includes a functional, comparative and phylogenetic toolbox, making use of reference proteomes from 319 eukaryotes, 1,678 bacteria, and 115 archaea. The TRAPID platform is available at: http://bioinformatics.psb.ugent.be/trapid_02.
Detailed information about the platform and its capabilities are provided in the different sections of this page. In addition, we provide step-by-step tutorials here, to guide non-experts through the different steps of processing a complete transcriptome using TRAPID.
Sample data, including a Panicum transcriptome (from Meyer et al., 2012) and several microbial eukaryote transcriptomes from the MMETSP (Keeling et al., 2014) can be found on TRAPID's public FTP.
Software requirements
All you need to use TRAPID is any modern browser with JavaScript enabled. TRAPID was tested using Firefox 76 & Chrome 81, on Ubuntu and Windows.
Citations
In case you publish results generated using TRAPID, please cite this paper:
TRAPID 2.0: a web application for taxonomic and functional analysis of de novo transcriptomes
Francois Bucchini, Andrea Del Cortona, Łukasz Kreft, Alexander Botzki, Michiel Van Bel, Klaas Vandepoele
Nucleic Acids Research, 01 July, 2021
In case you publish results generated by third-party resources used in TRAPID, please cite the appropriate papers as well. References can be found on the tools & parameters documentation page.
User authentication
Data security is a necessary concern when dealing with online platforms and services. TRAPID requires users to be registered, and no user can access the data of any other user, except if explicitly requested (shared experiment). User authentication is performed through username/password combination.
To create a TRAPID account, click on the
register button of the header of TRAPID's website. After filling in your information and registering, a password will be sent to you. Using the email address/password combination, the user gains access to the user-restricted area within the TRAPID platform. We recommend users to change their password after logging in for the first time (Account > Change password).
Step-by-step instructions on how to create an account and log in can be found in the tutorial .
Creating TRAPID experiments
In order to be processed, the transcriptome data must be first uploaded to TRAPID. Before doing this, it is important to note that a user has the ability to create different experiments for different transcriptome data sets, with a maximum of 20 experiments per user. So analyzing different transcriptome data sets at the same time is perfectly possible.
When creating an experiment, the most important setting to adjust is the reference database to use. The PLAZA reference databases should be very good for transcriptome data sets from plant or green algal species, while the eggNOG reference database should be used for any other species, such as animals, fungi, or bacteria.
An overview of the content of the various reference databases can be found in the tools & parameters documentation page, and step-by-step instructions on how to create an experiment can be found in the tutorial.
Uploading transcript sequences and job control
After a TRAPID experiment was created, the user can upload his
transcriptome data to the platform. The transcriptome data must be made available in FASTA format.
The maximum allowed size for an input file is 32MB. However, to accommodate for the rather large file size associated with plain-text multi-fasta files,
the uploaded file can also be compressed (.zip or .gz).
Input files may also be uploaded by providing a URL to a specific transcript
file (e.g. FTP site, cloud, public Dropbox URL, etc.); this option allows to upload larger files (max. size 300MB).
Please note that this option requires to provide a direct link to the data.
If the transcriptome data is split over several files, the user has the ability to
continue uploading data (via file upload or URLs) into his experiment before starting
the processing phase.
Once your sequences have been successfully uploaded into the database, an e-mail will be sent.
At any moment and during all processing steps (upload, initial processing, running downstream analyses from the web application), users can check the experiment's jobs or log, both accessible via links from the experiments overview page (outside an experiment) or the header (within an experiment).
- The job management page enables users to check the status of their jobs, and to delete them if needed. It also provides information about the status of TRAPID's underlying computing cluster.
- The experiment log stores detailed information about all the experiment's computation steps, used tools and parameters. It can be exported to a flat file.
processing or error, you can go to the experiment Status page and modify the status (e.g. to finished).
Step-by-step instructions on how to upload data can be found in the tutorial.
Performing transcript processing
The initial processing phase of the TRAPID platform is the next step, and necessary
before any of the user downstream analyses can be performed. This phase is initiated by clicking the Process transcripts
button on an experiment overview page. For this step, the user should consider
the options carefully, as they may seriously impact the downstream analyses.
Step-by-step instructions on how to process
transcripts can be found in the
tutorial.
Initial processing options
Similarity search database: whether either a single species or a phylogenetic clade will be used for the similarity search. A
single species is a good choice if in the reference database a close relative of the
transcriptome species is present. If a good encompassing phylogenetic clade is available, then
this is also a solid choice. By default, this is set to the most general clade of the reference database.
Note that if eggNOG 4.5 is used as reference database, it is not possible to choose a single species as similarity search database, due to the very high number of represented species.
Maximum E-value threshold: the maximum E-value cutoff to use for the DIAMOND similarity search.
Rfam clans: TRAPID identifies potential non-coding RNAs within the input data, using Infernal against a selection of Rfam RNA models. This option sets the Rfam clans to search for. By default, a collection of models corresponding to ubiquitous non-coding RNAs is used.
Gene Family Type: for PLAZA reference databases (PLAZA 4.5 monocots/dicots, pico-PLAZA
3.0, PLAZA diatoms 1.0), this is Gene families (TribeMCL clusters) or Integrative
Orthology. The latter may be selected only in case a single species was selected as Database Type. For eggNOG, only Gene families can be selected (which corresponds to eggNOG ortholog groups).
Functional annotation (only for PLAZA reference databases): the strategy used for functional annotation to be transferred from gene family to transcript. In general, Gene families is the most conservative approach
while Best hit is yielding a larger number of functionally annotated
transcripts. We recommend combining both methods and use Both (default), as it yields the
largest fraction of annotated transcripts and produces the most annotations.
Taxonomic scope (only for eggNOG): define eggNOG mapper's taxonomic level used for annotation. We recommend keeping the default value, Adjust automatically.
Perform taxonomic classification if checked, the similarity search is preceded by a taxonomic
classification of the transcripts, performed using Kaiju against the NCBI NR protein
database. The main purpose of this step is to enable the identification of potential contaminant sequences within single-species transcriptomes or to facilitate the analysis of transcriptome data from communities (or single-cell transcriptomes).
In addition, it is possible to define transcript subsets from the taxonomic classification
results, for quick sequence extraction or downstream analyses.
Input sequences are CDS: this option should be checked if input sequences are CDS (nucleotides). The ORF prediction step of the initial processing will be skipped and all sequences will be translated in +1 frame. Genetic code: the genetic code to use for ORF prediction and translation of input sequences. Any translation table from the NCBI taxonomy may be selected.
Click Run initial processing to launch the job. During this step, the experiment will become unavailable while the server performs
the initial processing of the data. Again, you will receive an e-mail when the processing has finished.
Initial processing time
The processing time depends on various factors. The most impactful are the size and composition of the input data set, the selected reference and similarity search databases, the number of selected Rfam clans, and the taxonomic classification. We can however give a few indicative numbers.
For the example Panicum data set (25,392 transcripts) discussed in the manuscript, the complete processing using PLAZA 4.5 monocots and all options set to their default value (no taxonomic classification) took around 1 hour and 3 minutes in total. The initial processing of a diatom-dominated phytoplankton community metatranscriptome consisting of 143,308 sequences took around 2 hours and 12 minutes in total, using Pico-PLAZA 3 as reference database, with all options set to their default value and performing taxonomic classification.
Basic analyses
After the initial processing of the data has finished, several new data types are available for the TRAPID experiment: gene families, RNA families, and functional annotation (GO terms, protein domains, or KEGG orthologs). Using these extra data types offers exciting new analyses to the user, accessible from the side menu of the experiment. Step-by-step instructions on how to explore some of these data types can be found in the tutorial.
General statistics
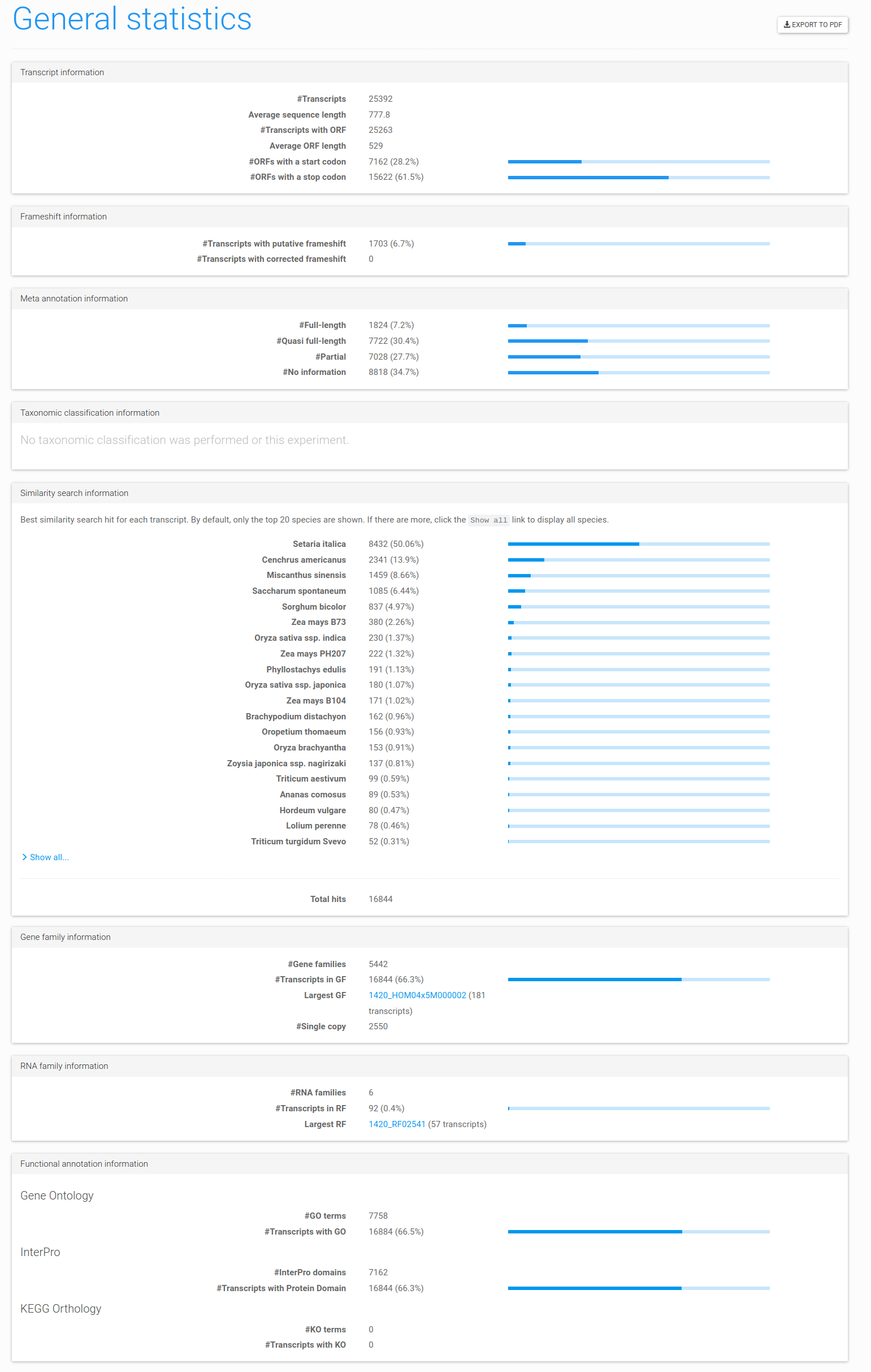
Figure 1: general statistics page example. Statistics generated using the Panicum example data set.
The general statistics page offers a complete overview of ORF finding, taxonomic classification, gene or RNA family
assignments, similarity search species information, meta-annotation and functional
information. This report can be exported to PDF by clicking the Export to PDF button in the top right.
The length distribution of the experiment's transcript and predicted ORF sequences can be inspected via the Sequence length distribution page.
Transcript subsets
Within a TRAPID experiment, transcript subsets can be defined from any arbitrary list of transcript identifiers. For instance, if the data set comprises transcriptome data from different sources (e.g. different species, tissues, developmental types or stress conditions), then the user has the ability to define transcript subsets corresponding to these sources. Subsets may either be uploaded as a file (from the Import data page, providing a list of transcript identifiers for the subset), or created interactively (for instance from the Taxonomic classification page, as shown in the dedicated section).
By creating transcript subsets, several new analyses become available, such as comparison of functional annotation between different subsets, or functional enrichment analysis. Note that it is possible for a transcript to be assigned to multiple subsets, and that three transcript subsets are defined by default after initial processing completion, corresponding to protein-coding transcripts, RNA transcripts, and ambiguous transcripts (transcripts assigned to both an RNA and protein-coding gene family).
Step-by-step instructions on how to define and analyze subsets can be found in the tutorial.
Searching for data
The user has the ability to search for various data types within their experiment, using the search box present in the header of the experiment. Functional annotation (GO terms, protein domains, or KO terms) can be searched using either identifiers (e.g. GO:0005509) or
descriptions (e.g. Calcium ion binding). Searching functional annotations by identifier looks up exact matches only, whereas searching by description yields all results overlapping the query.
Similarly, gene and RNA families can be searched using either TRAPID's internal identifier (with a exp_id_ prefix) or
the reference DB gene family identifier. Finally, the search box is convenient to retrieve the transcripts having a given meta-annotation.
Exporting data
The TRAPID platform allows the export of both the original data and the annotated and processed data of a user experiment. This data access is available under the Export data header on an experiment page and includes structural ORF information, transcript/ORF/protein sequences, taxonomic classification, gene/RNA family information, and functional GO/InterPro/KO information.
The remainder of this section consists of a description of each type of export file, organized by category, complemented by minimal examples (ten first records). Please click on the Toggle example links to show the corresponding minimal example export file.
Structural data
The structural data export file is a tab-delimited file providing the following information for each sequence of an experiment:
Transcript identifier: the transcript sequence identifier.Frame information: the detected frame, strand, and full frame information (homology support) for the inferred ORF sequence of the transcript.Frameshift information: flag putative frameshifts (0/1 boolean value).ORF information: the start/stop coordinates of the inferred ORF sequence and the presence of start/stop codons.Meta annotation: the meta-annotation complemented by meta-annotation scoring information.
#transcript_id detected_frame detected_strand full_frame_info putative_frameshift is_frame_corrected orf_start orf_stop orf_contains_start_codon orf_contains_stop_codon meta_annotation meta_annotation_score contig15600 3 - hit="Seita.5G133300" 0 0 0 1417 0 1 Quasi Full Length std_dev=562;avg=1615;orf_length=1416;cutoff=491 contig14583 2 + hit="LOC_Os12g34104" 0 0 799 945 1 1 No Information null contig14334 3 - hit="Seita.9G362500" 0 0 0 2689 0 1 Quasi Full Length std_dev=540;avg=2737;orf_length=2688;cutoff=1657 contig15854 1 - hit="Seita.3G183900" 0 0 0 1517 0 1 Quasi Full Length std_dev=762;avg=1912;orf_length=1518;cutoff=388 contig15185 3 - hit="Sobic.004G069000" 0 0 0 2047 0 1 Quasi Full Length std_dev=312;avg=2228;orf_length=2046;cutoff=1604 contig14563 1 - hit="OB08G26510";alternative_frames="-2" 1 0 1365 1832 1 1 Full Length std_dev=616;avg=1673;orf_length=468;cutoff=441 contig14653 1 - hit="Seita.3G037300";alternative_frames="-2" 1 0 957 2204 1 1 Full Length std_dev=373;avg=1181;orf_length=1248;cutoff=435 contig15055 1 - hit="Seita.1G378200";alternative_frames="-3" 1 0 0 1847 0 1 Quasi Full Length std_dev=321;avg=1837;orf_length=1848;cutoff=1195 contig15538 3 + hit="Sobic.001G116500" 0 0 0 1693 0 1 Quasi Full Length std_dev=455;avg=1572;orf_length=1692;cutoff=662
Taxonomic classification
The taxonomic classification export file is a tab-delimited file that provides, for each transcript of an experiment, their associated taxonomic label (NCBI tax ID of the lowest common ancestor, set to 0 if a transcript was not classified). In case a transcript was classified, classification metrics (score, number of matching tax IDs, number of matching sequences) and full taxonomic lineage are also provided. The classification score corresponds to the length of the best MEM sequence found by Kaiju. Toggle example
#counter transcript_id tax_id score n_match_tax n_match_seqs lineage 1 contig00001 206008 263 15 22 Panicum hallii; Panicum sect. Panicum; Panicum; Panicinae; Paniceae; Panicodae; Panicoideae; PACMAD clade; Poaceae; Poales; commelinids; Petrosaviidae; Liliopsida; Mesangiospermae; Magnoliophyta; Spermatophyta; Euphyllophyta; Tracheophyta; Embryophyta; Streptophytina; Streptophyta; Viridiplantae; Eukaryota; cellular organisms 2 contig00002 0 3 contig00003 206008 387 7 22 Panicum hallii; Panicum sect. Panicum; Panicum; Panicinae; Paniceae; Panicodae; Panicoideae; PACMAD clade; Poaceae; Poales; commelinids; Petrosaviidae; Liliopsida; Mesangiospermae; Magnoliophyta; Spermatophyta; Euphyllophyta; Tracheophyta; Embryophyta; Streptophytina; Streptophyta; Viridiplantae; Eukaryota; cellular organisms 4 contig00004 206008 673 7 11 Panicum hallii; Panicum sect. Panicum; Panicum; Panicinae; Paniceae; Panicodae; Panicoideae; PACMAD clade; Poaceae; Poales; commelinids; Petrosaviidae; Liliopsida; Mesangiospermae; Magnoliophyta; Spermatophyta; Euphyllophyta; Tracheophyta; Embryophyta; Streptophytina; Streptophyta; Viridiplantae; Eukaryota; cellular organisms 5 contig00005 0 6 contig00006 206008 107 7 22 Panicum hallii; Panicum sect. Panicum; Panicum; Panicinae; Paniceae; Panicodae; Panicoideae; PACMAD clade; Poaceae; Poales; commelinids; Petrosaviidae; Liliopsida; Mesangiospermae; Magnoliophyta; Spermatophyta; Euphyllophyta; Tracheophyta; Embryophyta; Streptophytina; Streptophyta; Viridiplantae; Eukaryota; cellular organisms 7 contig00007 2014292 11 8 11 bacterium (Candidatus Ratteibacteria) CG23_combo_of_CG06-09_8_20_14_all_48_7; unclassified Bacteria (miscellaneous); unclassified Bacteria; Bacteria; cellular organisms 8 contig00008 206008 74 15 44 Panicum hallii; Panicum sect. Panicum; Panicum; Panicinae; Paniceae; Panicodae; Panicoideae; PACMAD clade; Poaceae; Poales; commelinids; Petrosaviidae; Liliopsida; Mesangiospermae; Magnoliophyta; Spermatophyta; Euphyllophyta; Tracheophyta; Embryophyta; Streptophytina; Streptophyta; Viridiplantae; Eukaryota; cellular organisms 9 contig00009 206008 332 7 22 Panicum hallii; Panicum sect. Panicum; Panicum; Panicinae; Paniceae; Panicodae; Panicoideae; PACMAD clade; Poaceae; Poales; commelinids; Petrosaviidae; Liliopsida; Mesangiospermae; Magnoliophyta; Spermatophyta; Euphyllophyta; Tracheophyta; Embryophyta; Streptophytina; Streptophyta; Viridiplantae; Eukaryota; cellular organisms
Gene family data
Three types of gene family data export files are available:
-
Transcripts with GF: a tab-delimited file that contains the transcripts of an experiment and their associated gene family (if any). Toggle example -
GF with transcripts: a tab-delimited file that contains, for each gene family of an experiment, the number and identifiers of transcripts assigned to the gene family (on a single line). Toggle example -
GF reference data: a tab-delimited file that contains the reference data (GF name and members from the reference database) for each gene family of an experiment. Toggle example
#counter transcript_id gf_id 1 contig15600 325_HOM04M000078 2 contig14583 325_HOM04M024306 3 contig14334 325_HOM04M002301 4 contig15854 325_HOM04M000265 5 contig15185 325_HOM04M007940 6 contig14563 325_HOM04M000016 7 contig14653 325_HOM04M000527 8 contig15055 325_HOM04M002035 9 contig15538 325_HOM04M001852 10 contig15718 325_HOM04M000525
#counter gf_id transcript_count transcripts 1 325_HOM04M000288 1 contig21531 2 325_HOM04M000289 8 contig14024 contig13643 contig15820 contig14435 contig00084 contig24153 contig14741 contig25148 3 325_HOM04M000290 4 contig12096 contig12098 contig24795 contig21293 4 325_HOM04M000291 8 contig08387 contig24777 contig20960 contig19804 contig17222 contig22643 contig14830 contig19514 5 325_HOM04M000292 11 contig19081 contig18701 contig06441 contig06440 contig22975 contig06437 contig19799 contig10181 contig06438 contig24751 contig21978 6 325_HOM04M000293 1 contig18090 7 325_HOM04M000294 1 contig16262 8 325_HOM04M000295 2 contig17068 contig21683 9 325_HOM04M000297 2 contig20335 contig06599 10 325_HOM04M000299 11 contig05344 contig22288 contig05342 contig20433 contig05343 contig16646 contig17772 contig11019 contig23477 contig11020 contig15118
#counter trapid_gf_id reference_gf_id gene_id 1 325_HOM04M002770 HOM04M002770 Pp3c2_32420 2 325_HOM04M002770 HOM04M002770 TAE37408G001 3 325_HOM04M002770 HOM04M002770 TAE37408G002 4 325_HOM04M002770 HOM04M002770 ATR0680G209 5 325_HOM04M002770 HOM04M002770 GSVIVG01020946001 6 325_HOM04M002770 HOM04M002770 PH01000860G0510 7 325_HOM04M002770 HOM04M002770 TAE27601G003 8 325_HOM04M002770 HOM04M002770 PAB00003424 9 325_HOM04M002770 HOM04M002770 Solyc03g025590.2 10 325_HOM04M002770 HOM04M002770 PAB00037282
RNA family data
The export files for RNA family data are identical to the gene family data export files (but containing RNA family information). However, no export file for reference information of RNA families is available, as this data is not stored anywhere within TRAPID. Please visit the Rfam website to retrieve this information.
Sequences
Sequence export files are FASTA files for a chosen type of sequence and a selection of transcript sequences from an experiment. Exported sequences can either be the uploaded transcript sequences, the inferred ORF sequences, or amino acid (translated ORF) sequences. It is possible to export sequences for all the transcripts of an experiment (default) or for any defined transcript subset. Toggle example
>contig15600 AAATTGGAGAGGCAGCTAGCTTCTTCTCAAGTTCCCAAGGAGCAGCAGATCAATTTAATAAAAGATCTGGAGAGGAAGGAAACTGAATACATGCGACTAAAGAGGCACAAAATTTGTGTGGATGACTTTGAGCTGCTCACTATCATTGGGAGAGGCGCGTTTGGAGAGGTTCGACTTTGCCGAGAGAAGACTTCTGGCAACATTTATGCAATGAAAAAACTCAAGAAGTCTGATATGGTTGTCAGGGGCCAAGTTGAACATGTTAGAGCCGAAAGAAACTTGCTGGCTGAAGTGGCTAGTCACTGCATTGTGAAGCTTTACTATTCTTTCCAAGACGCTGAGTACCTTTACCTTATTATGGAGTACCTCCCTGGTGGTGATATTATGACCCTTCTCATGAGAGAGGATACCTTGACTGAGCATGTGGCGCGATTCTATATTGCAGAAACAATTCTTGCTATTGAATCCATCCATAAGCATAACTACATCCACAGAGATATTAAGCCTGACAACCTGCTTCTAGATAAGAATGGTCACATGAAGTTGTCAGATTTTGGGCTGTGCAAGCCAATTGACTGTTCTAAGCTCTCAACCTTGAATGAAGATGAACCCATGGGCGATGACAATCTAAGGGAATCAATGGATGTTGACAGTTCTCTGTCTGACACAGCAAACGGTAGAAGATGGAGAAGTCAACATGAACAACTTCAGCACTGGCAGATGAACAGGAGAAAATTGGCATTCTCAACTGTTGGGACACCAGACTATATTGCTCCAGAGGTTCTGCTAAAGAAGGGATATGGAATGGAATGTGACTGGTGGTCCTTGGGAGCTATCATGTATGAGATGCTTGTTGGGTATCCACCATTTTATGCCGATGATCCAATAACTACATGCCGAAAGATTGTGCACTGGAGAAACCATTTGAAGTTTCCTGAGGATGCAAGGTTGTCAAATGAAGCAAGAGATCTCATTTGCCGGTTATTATGTGATGTTGACCACAGGATTGGCAGTTCAGGGGCAGATCAAATAAAGGCACATCCTTGGTTCCGAGGAGTTGCATGGGATAAACTTTATGAAATGGAAGCAGCATTTAAGCCTCAAGTAAATGATGAATTGGATACACAGAATTTCATGAAATTTGAGGAATTGGAAAATCCTCCAGCTAGAACAGGCTCTGGGCCCTCAAGAAAGATGATGCTAAACTCCAAAGATCTGAGCTTTGTGGGGTATACATACAAAAACTTTGATGCCGTGAAAGCAATAAAAATTTCAGATCTGCAAAGGAATTCATCTCTAACAAGGCCCTCCATTGGTTCGATATTCGGTCCACCAGGCATGGATTCTCCTATGGAAGCAAACGGAAGGGACACACATATGCACACAGTTTCATCTGGTGATCCAATGATTCCCTAA >contig14583 ATGGTTGTTGAAGAAGAGCTTGTACGGGCTCCGAGGCAGTGGTCAAAGAAATTCGACAAGTTCATGGTCGATCGATACCTTTCCATCCCTGTTGTGGGGGCAGATGAGCCTCCCTCTGTTCTTGCAGCTGCCTTAGGAGGTCTTTAG >contig14334 AAGAGAGACTTTGACCGGCAGATGGGGTGCATGGCCGGCGTGTTCCAGATCTTCGACCGTCGCCGCCTTCTCACTGGCCGACAACGTGGCGGCAGCCCTGGGACCGGGAACGAACTGCCTTCAGGCCATGATCTTCCAGGCAGCAGCATTTATGCCCCGGTGTATAATTCAGCTCATCCAAACACCACTCCGGAGAAATCGTTCAGCAAAAGTACGACGGAGAACAGTATCCTCTCAATGGAATCATCGAGGGTATCTTCATCTTCGTCATCCTGCTCTTCATTATCATCCCTGGACGGCAGCAAACCAGTACAGCAGGAGCTCCCTTACATCAACGAGGAGCTTGTTGAGGGAAGGACAGTGAGGAACTCACGAAGCTTAAAGTCCTCCAATAAAGTGGTTAAATCTAAACAGAGAAATACTGATTTTAGAGATGTCGTAAAGGACTCAATAAGCCGGGACTCTGGCGTCCTAACCATCAAGACCACAACGATGGCGCAGAGAAATGGATTGCACAAAGACTCACCAAGGCCGCTGCTGATATCCAAATCAACAGATGGAACTTACGTGATTGCGATCGATAGGAGCAGTGGGCTTCCAGCATATGTTGGTGAACCTAGCAGGCAGCCACGTTTCTCATGTGATGATCGACAGATGCTGCAGCAGGCAGAAGCTCAAGATAGCCAGATGCCTTCATCAAAGCTCAGGGAGCTTCCTAGGTTGTCCTTGGACAGTACGAAAGAGTCTGTCAGGCCAAGTTCGCATCTGAATGACTTTGGTTATGCAAAAGCCGATGACAGCCTCATTGATAATTTGAAGAGCCAAGCATCCCCGGGCCATCGACGGGCAAGCAGTGTCATTGCCAAACTCATGGGGTTGGAAGAGACTCTAGATAGCTCTGGGCCTGCAAGATCACACAGACAAGCACATGACATTCAAAATGGTCACCCGTCACAAATCCCCAGGAGTATCTGCCCTGATTCCAGTGTATCACAGCCGATGGTTCAGCCTCCTATACTGAAAACCTGTGAAGTCAGATTTGTCCCTGAAGCTGCCCCTTTGAAGCAGCGGGAGAGAGGTATAACAAGATACAATGATGAAGCAAGGCCAAGATCTGCATCCACCTATAATGACATGGAGAGAAGGCTCAGGCATCTCGCATTATCTGAATGCAATAAGGACTTGAGGGCTCTCAGGATACTTGGCAATTTGCACGCAAAGCACACACCATTCGAGAGAGATTACAATGCCAGATTATTGCCCATTCAGAAGGCAACCGCTGAAGGAAACAACACTACTGCTCAAGATCTCCAGTCTCCAGTTGTGATCATTAAACCGGCAAGAGGAATCATGAGACCAAATGCTTCAGTTGCTTCCCTTGCAGGACCAAAAGTCCACAGAAAGTTGCAGCATGAAGAGCGTCCTTTCACCAGGAAGAGTGACAACAGTGACAGGAAGAAGACTCATCCTCACCATGGAAGAGTTCATTCTAGGGCAGAGGAAGCTGTTGGCAGCACAAACTCACCATCGCCTTCTAGATTGTTGAGCCCAAGACTTGTTCAGAAGAAGTCAGACTGTGGAAGGATCCCTCGCTTGTCAGTCCCACCAATGTCTCCAGGCAAGACACCCGATGAAGTAGTGTCCCCAAGAGTCAGACTAAGGGCAAGGGCTGCACAAGCAAATAATATCTGCCGTGATGATAAGATGTCAATGATTCCTGAAAGTAGAATTAGTTTATCCAAGCATGTTGATATGGGCATTATCGATTATCCAAATCTTAATGTCAACACATCATGCAGCCATCAAAGCAATACAGTTTCAAAGTTGAATAATGAGGAACCACCACCAATCCTATCATCTAACAAGAAAAACATCCATCCACTGGAGAACATACCAAGTCCTATATCAGTCCTTGATGCCATGTTCTGTCAAGATGGGTTGTCACCTTCTTTGAGGAACATATCAAATTCTTTCCAAGATGTTTCGACACATACACTGGATGAGTGCTGGAACCCAGTAAGCCTCCCTGACACACCAATATTGAAGAAAAATTGTGAGGGTGACCATAAACTACCAGAAAACATGACAGCCCTTATCCAAAAGCTTGAGCTCTTGCAATTGTTGAGTGATGAGGCTCCAAGCACAAATGACAATTTGTTAATGGACACTGCCAATAAAGACCGCCATTACATTTATGAGATACTCTCAGCATCAGGCCTCCTGCACAGTGAACTAGGCTCCAGGATGATGCCCTGTCAGTTCCAGCTGCCAAGCTACCCAATCAACACCGGGGTTTTCCTTATCCTTGAGCAAGCAAAACCAGCCGCAGGAAAGCTTCACCGCAAGCTCATTTTCGATCTTACAAATGAGCTCATTGCTCAGAAAATACGCAATGGTGGTTCAGTGAGACAACCACTGCAGTTGTTTCGATGTAAGAAATCAAGTGGATTGCATCTTTTCAAGGAATTATGCTCAGAGATTGAAATACTCCAATCTGAGGCCTCAATAATAAGACTCTCTGAAGAGGAAGAGGAAGAGAGCAAGCCGGCAAAGAATGCAGTAGATGAAATGGGAAAATGGAAGAGTTTTGACAGCGAGCTACAAGGGATGGTTCTGGACATTGAAAGATACATCTTTAAGAATCTCATTGACGAGGTAATAAATGGTGAAGCCATGAGAAAGGTGTAA >contig15854 CGGCTACCTGCGGCAGTTGCCAATGGAAACTGTGGTGAGGAGGAGGGCCGGGGGCAGGGTATGTGTATGATTATGAGGGAGACATTTGGTGGGGCGCTGCTCAGTCAGGTGAAGTGTCTTTTGTGCAAGGGCGAGTCAAACAAGACAGACGATATAATGGATATCAGTCTCGATCTACCTGGGAGCAGTTCTGTTGCTGATGCCCTTGCCCGCTTCTTTCAGCCGGAGATCTTGGAGGGTGCAAACAAATATAGTTGCGAAAGATGTAAAAAGCTCACGTCAGCACGGAAGCAAATGTTTATACTCAGGGCACCTAAGGTGCTTGTCATACAACTCAAGAGGTTTGAGGGGATAAATGGTGGAAAGATCAACAGGAGTATAGAATTCAAGGAGGCTCTTGTTCTTTCTGACTTCATGTATGATAAAAACCAGGATTCACATCCAGCATATAATCTTTTTGGTTGTATCGTCCACTCAGGGCTCTCCCCTGATTCTGGTCACTACTATGCGTATGTTAAGGATGCTATTGGTCGATGGTTTTGTTGTAACGATTCTCATGTCTCCCTATCAAGCTCTCAGAATGTTTTGTCTGAGAAGGTCTATATCCTATTTTATATACTGAATTCGAAAAATCAAAAGCCTAGTCCAAATGGTTGCTCTTCTACTGCAGCTAAACCTTTCAGCACCAATGGAAGTGGCATATCAAGCACCTCACCTAATGAGACCTTGAAGATACCTTTGATAAAACAGAATGGTTCATGTTCTACCGAAGGTAATGCCCTGCTGCCCCTGAAGAATGGCAAGATTGCATCAGGTCCACTTATCAAGCCGATTCACTTTAAGAATAGTGGGACAGAGAAGGTTAAGTCAAATGGCAAAGAAAACCTCCCTTCAAAAATGAACCCAGAAGTTAATGAGAGTGCTATACCATCAGAATCAAATGGGCACAAGACTGGAAAATTTGCGGAACCCAGTAAAAAGGATGCCGATGGTACTATGTCTTGTGGAAAGATTGATGATCATTCAGAAAGAATATTGCAGGATGCAAATGGAAATGGCCACCTGATTCGTTCCCAATATCTTGGCGAGGCCAGTAATGGTAATGCCACATGTGCTCAACAGTATTCTGAGCAGTCATCTGGTGCTGTTGCCTCTAAAAGTCCAGTTTCACACCATGAAGAGTCTGCTAAAGTAAAGGATGTGGCGAATTCATCCAAGGATAGCGTGCACCTGAAACGCCAACATGAAGAAGACAGGTTTAAGGAAGTGCTTGCTAAGTCAGCTTCTTCTGAGCTTCGGCTGTCTGTTTGGGTGGATGATGTCAGTAATTTTATGCGCTCACAAAAGAGACGGCGAATTCAAAACCCAGGCATTCCTCAAGATATTGATGCAATGAGAAAGCTGCTAAAATCAGATTCTGAAAGAATATTTAGGTCAAAAGTTCCGGAGTCACTGGTGGAAAGTCTCATTCAACGCCTGAGATCATACTTTGAGAGCATATATCCACCAAATCCTTAA >contig15185 CCAAGTGCTTCTCAGGATAGGGAGCTGAGCATGAGCATCGAGGTCCTGAAGACTGAATTTGAGGCTGCCTTGTCTATCCTAAGGAAAAAAGAGAGGGACCTTCGGGATGCGGAGAAGAAAGTTTCTGTTGATCGGTCAAGGCTGAACCAGACGAAGCAGGACCTTGATCAGAGGGAGGAAGACATCATCAAAGCGTATTCAAGACAACATGAAATGGAGAAAGCACTGATGAAGGCGAGTAGGGATTTATCTTTACAGGTTAGGCAGATCAATAATCTGAAGCTTCTAGTTGAGGAACAAGACAAGAAAATTGTTAGTTCACAAGCTGCGCTTTCTAAGAAGGTGATTGAAGTGGATAAGCTTAAACAGGATATGCTGAAGAAGAACGAGGAAGCAGCCTTGATGCGTTCAGAGATTGAGTCCAAGGAACAAGAGCTTCTTGTAGCTAATCAGGCCCTTGCACGGCAAGAAGCAACAATTAGGGAGCTCCAGAATGAAATTAAAACAAAGGAAACTGAGGTTGCCAGATCAAATGAATTGAGGAAAGCTAATGAGGAGAAACTGAAAGTTGCAGAACAGGAACTTGAGAAGCAGAGTTTAGGATGGATAGCAGCACAGCAAGAGTTAAAGGAACTGGCGCAAATGGCATCCAAGGATAAGGATAATATCAAGGATACTATAGATGACTTCAAACGTGTGAGATCTTTGCTGGATGCTGTACGTTCCGAACTAATGGCTTCAAAAGAGGCTTTCACCTTCTCGCGCAAGCAAATTGAAGACCAAGCGGCGCAGTTGAGTAAGCAAGTGCAGGAACTCACAGACCAAAAAGCATTGCTTATTTCTTATACTCAGAGTCTGGAAGCTGCTCAACTGGAGATTCAAGGAAAGACAAATGAGCTCAGCGCTGTAAAAACTCGCTGTAGTGAGCTTGAATCACGGTTACTTGAGGAAATGAAGAAGGTTGAGTCCCTAGAGGCTATGTTAACAAAAGAAAGGGAGAGCTTGGAACAGAAAACTAAGGAGGTAGACTTGCTTCAAGAGGAGCTAGCTCGGAAGGAGAATGAGTACTTCAATTCACAAAAGCTTGTCGAAACAAAAGAGAATGAGTTGTTAGAGGCCAGACATGAAGTCGAAGATATGAAATTGAAGGTGGACTCCATACAATTTGCCGTTCAAGAGAAAGATTTGGAGCTTCTGGAGACACAACGAAGACTTGACGAAGTTAACAATGAAGTTGTTGAACTTCAGCATATGATAAATACCAAGGAGGATCAACTTGTTCAGGTTAGATCCGAACTACAGGATAAAGAGCAACGTATACAATTAATGCAGGATGAATTGGATAAGATGAGATTAGGACGCTCACAAGCTGAATCTGTGGTGCAAAAGATAGTTGAGCTCACTGGCAATCTTATAGGTTCTGTAGAAGGTGAAGAATTTGACATTTATAACTTGCTCGATGATGAAATTTTAAGCACAGACACAGCCCTTGAGTCGAATTTGCATAAACATAATCAACTGGAGGCTGACATCGACATGCTGAAAGAATCCTTGGAACAGAAAGACGTGGACTTAAGAGCTGCATATAAAGCACTTGACGCAAAAGACCTAGAGCTGAAGGCAGTACTTAGAAGGTTAGATGTGAGGGACAAGGAACTAGACAAATTGGAAGAGCTATCCATAGATCCCAACGACGTCAGGAGACTGTCTAGCCTTGCTGATGAGGCAACCAAAGCCAACAATGTGGAAGAAGTGGAGCTCCGAAAGCATGAACTCGAATCTGTGGAGGGGGAGGCACTAGCTGCTAGTACTATGTTGAAGAAGCTTGCCGATATCACTAAAGCATTCTTGAGAAGTGGTAGAACCGATTCTGGTACCAATCTGCTTGCATCCCGAAATGCAAACATCAGTGAAGGTGCTTCCAAAATGGAACCAAAGAAGAAAATGAACGTGATTCTTGAAGCTAAAAAGGAGATCGTTGGGCTGTTTTCTTTGACAGAAGAACTCGTCGCCAGTGCTGGAATAAATGACGCTGAGGAACCGTAG >contig14563 ATGATTTTCAGCTTAGAAGAGCTTGAAAAGGCCACAAACAGTTTCGATGAATCTCGAAAACTTGGTGGTGGGGGCCATGGCACTGTGTACAAAGGTATTTTGTCTGACCAACGACTCGTTGCTATTAAAAGGTCAAGGCATGCGATTAAGAGAGAAATTGATGATTTTATCAATGAGGTCGCCATACTTTCTCAAGTAAACCACAGGAATGTAGTGAAGCTCTTTGGATGTTGTCTTGAGACAGAAGTTCCATTGCTAATCTATGAGTTCATTTCGAATGGGACACTTCATGACCATCTACATGTATGTGCTCCACAATCACTATCATGGAAGGAGCGAATGAGGATAGCCCTCGAAATTGCAAGATCTCTTGCTTATTTGCATTCTGCTGCTTCAGTTGCGATAATCCATAGAGACATAAAGACTACCAACATACTTCTTGATGATCGGTTGATAGCAAAAATATAA >contig14653 ATGGCTGCCTGGTCTGAACTGAAGCAATTGCCCCGGGTAGGTGAGCCATCGTCCTCATCATGTCCACTCGATCAGGATGATGAGGAGCATGAGCAGGCCATAGTTACTCGCACTCTTGCAAGCTTGAACCAGGCAAATGGTGGCAAGATGCCACATCAAAAAGAAAAACAGCAAAGCAATAACCGTCCATCATCACGAAGATCTTATCCTAAATCAAATCCTTCATTCTACCGATCACATTTACCAAACCAGGCTTATCCTAGTGTTCCACCGGAGCAAGCAATGTACCATATGTGGCACCGGGTGCAAGCAACACAACCAGCACCTAGTTTTCCAATGGTACCAACTATGGGTAATACGAGGTTCCAACCACCAGCAGCCATGCTCTCCATGTACTCTTCACCTCGAGGGCAATTGGCCACACCGGCCTGCCAAGATGCACTAGGTCTTCTTCCATGTTTTCCTGAAGGTGCTCCTGCCCTCCCAAGGTACTTCTCGCCTTACCCTGTCTCCTATGTGCCAAGAAGTCCTGTGCCAGCTACTGTTCACAAAATTCATGAGAGAAGGCAACATCTTGCTGAAACAGTTGAGCTTCCTGATGCCGCGGTCTTCTCCCAGTATGGTGGTCCACACAAGTTTCAAGAACCACCCAAAAATGGGAAAGAAGACCGTACTGGTAGTAGTGCTTCCCCTGAGAAAGAAATTATTGCTCCTCTCAGTATCTCAGGCTCAACCACACATCCATCATCGCCAAAGTTGGATCTGAATGAAGAAAAAGTAACTTTGGGCAGTAAGCCAAATAAGTCCCAAGAACAACAGCCAAAGTCACCACCTTGGGTTAGCCCTTCAATTCCAGCACACGGTTCTATCCAGGGAAAGCATTACACTAGCTCTGTTCAGCATGATGAGCCCATCCACAGAAATGATCCACCTCAAACGAGTCGGCCTTCGTTGCCAGAGTTGTGGTCTTCCTGCTCAGCAGCTGCACCCAGATCTGGAGCTGGAGCTGCAGTCCCTGTACATTCACCAGGTCCTGTATACCAGCAGCGACCTCCTTGGCTGGCTGCCCCTGTTACAGTCAGAACCGCCATCCCAGTGTGTTCAGCTAGGCCGAATGCGGTGAATACAGCTGGTGGAGCAGCTCGGGTAAGACCCATCACCCAGAACCGCTTGGCCCTGGCTAGAGGTGAGCCAGAAACTCCCAGAAATACGAATAATGGGGAAAGAGCCCTGAACAGCGAGCACTGA >contig15055 CAACAACTCCACGGGCTCCTCAACAGCCTGCACAGCAAGTTTCTGTTCCAGCTCAAGCCCATGGCTCTATTCCTGCCAAGCAGGGTGTTCCGTGCTATTACTTTCAGAAAGGCATGTGCGACAAAAGGGGACAGGTGTGCCTTCTCGCACGGCCCACAGCCTGCTGGGAACCCTGCTCCACAGCCGCCACCGGCTGCCAAGATCTTTGCTCCTGCTTTGCAGCCTAATTCTCAATTGAAGAACTCGTGGACAAAACCCAACTCTTCAGGCCAACAGAATACACCTGCAGGCATACCAGACAAGTCAAAGCTTAGCACTCATGATGCTAAACCAGTTCAGAAGCAACATGTGACGAGTAGGGTTGACCACTCGTCAAGAACTTATCAGAATCACAGTAATTCTTATGCGCAGTCTGGTACAACAAAGCATTACCAACCTCAGCCTTCAGTCCAAGATGGCTTAAATGAGAATGGTATGGAGGCGGGTGAATTTGTGAGGGAACCTTCCGCTGGTTCTGGTGTTCTTGTTGGTGGTGTCGATGATGACTCTGAACAGTCATTCAAGGGGAACCGTAGCAGCTACCACCATCATACTTCACATGGTGGCTATGCACCAGAAAGGTCATACAGAAGTTCAGCTGAGAGATTGTCATCAGACAAAAGGATCTCGGAGCAAGAACCTATGCCTGCTGTGATTGCTGGCAGTTCAGATTTGCGCCACAGGTTACTGAAGCAAAGAAGGCTCAATAATAATTTAGGATCAACTGGGGCCCCTGATATGAATGACTCGTGTCTTGAGGGTGAACGCAATGACCAACGTCGCTGGAGAGGTGAAGAGCATGATGGCTCCCTCCCCCGATCTCGATTGCGTGATAGAATAAGACTGCCTGGAGCGACATCCTTTGACAGACATGGATCACGCTCAGAGAAGGAGTGGGATAGAGCGCCTAGAGGCAGGCTGTCACCACCGGAACATTCGGATCTTCGAGGGAAGCTTCATGAAAGGCTAAAGGTCAGATCAGCTGAGGAGATACCGGTTAATAGTGTTAAGAGCTCGGTGGTGAAGGCAAGTAGTGGTGAGGATGCTGAATCATTGAACTTTGCTGGTCCAAAGAGCCTTGCCGAGTTGAAAGCAAAGAAGGGTGGTGCCAGCTCATCACAGGGAGAGGCCATCGTCAAGGGTGTGGGCTTATCTCGTGTGACTTCAGGAATAATTTCGAGCAGGGAGCCTGCTCCATTTGAAGGTCCAAAGCCACTAAGTGCCATACTGAAGAGAAAAAGAGAGGTGGCCAGTGAGGACGCTGCTGCCCATTTTGGCAGCATACAGGAAGAGGATAATGCTGCAGGAGTGGACGAAGAATCCCAGATCTTAGCGGATGACACGGTTGAGGAGAACATGGAGGGAAATACAGCAGCAGAAGAAGAAGGGGAAGAGGAAGCCTTCCATCCTGAAGATGATGTAGCGTATGACGACAACGCCGATGAAGCTACAGGTCAAGAGCTGGAGGAGCATCAAGATGTAGAGACAGCAGGGGAAGGGGAAGACTCTGACTACGAGGCGGCTGACACCAACGCTGCTGCAGGTCAAGAGCTGGAGGGGCATCAGGATGCAGAGGCAGTAACCGAAGACTATGACTACGAGGCGGCTGACGCCAACGCCAACTCCAACGCCGCGGCAGGTCAAGATCTTCAGGATGTAGAGGCAGCAGCCGAAGACTATGACTATGAGGCGGCTGACGTCAATGCCGAGGAGGATAATGAGTACCATGAGTACCAGGATGATGATGATGATTTGGAGGATGACGACGATGATGATTTTGCGCGGAAAGTGGGCGTGATGATCTCCTGA >contig15538 TCCGGCCTCCGGAGGAAGGTTACTGTATTCCAGCAACCTCATTATCTACAGAACTTTGTCCAATCAACATTCAATGCCCTTCCTGCTGAAGAAGTAAAAGGTGCGACCATTGTTGTCTCTGGTGATGGCCGCTATTTCTCAAAAGATGCTGTTCAGATCATAACAAAAATGGCTGCTGCCAATGGAGCAAGACGTGTTTGGGTTGGGCTAAACAGTCTCATGTCTACTCCTGCCGTTTCTGCTGTCATCCGTGAAAGAGTTGGTGCAGATGGATCAAAGGCTACGGGTGCCTTCATCTTAACAGCTAGCCACAACCCAGGTGGTCCGACTGAGGACTTCGGGATCAAATACAATATGGGAAATGGTGGGCCTGCCCCTGAGTCTGTTACTGACAAGATTTTCTCTAATACAACGACTATCTCTGAATACCTCATCGCCGAAGACCTTCCAGATGTTGATATTTCTGTTGTTGGTGTCACTAGCTTCAGTGGACCTGAAGGCCCCTTTGATGTGGATGTCTTTGACTCCAGTGTAGATTACATAAAGTTAATGAAGACAATTTTTGACTTCGAGGCAATAAAGAAGTTGCTGAGCTCTCCAAAATTTACATTCTGTTATGATGCCCTCCATGGTGTTGCTGGAGCTTATGCCAAACACATCTTTGTGGAAGAGCTTGGTGCTGATGAAAGCGCTCTGTTGAATTGCGTCCCAAAGGAGGACTTTGGAGGTGGCCATCCGGATCCTAACCTCACCTATGCAAAAGAGTTGGTTGAACGGATGGGTCTTGGAAAGTCATCCTCAAACGTCGAACCTCCTGAGTTTGGTGCAGCTGCTGATGGAGATGCTGACCGAAACATGATTCTGGGTAAAAGATTCTTTGTGACGCCATCGGACTCTGTTGCCATTATCGCGGCCAATGCTGTTCAGTCAATTCCTTACTTTGCTTCTGGCCTGAAGGGAGTTGCAAGGAGCATGCCAACATCTGCTGCCCTTGATGTTGTTGCGAAGAATTTAAATCTCAAGTTCTTTGAGGTGCCTACTGGGTGGAAATTTTTTGGCAACTTGATGGATGCCGGAATGTGCTCAATCTGTGGTGAAGAGAGCTTTGGCACCGGTTCTGATCACATTCGTGAGAAGGATGGTATTTGGGCTGTGCTTGCATGGCTGTCTATTCTTGCTTTCAAGAACAAGGACAACCTTGAAGGAGATAATAAGCTTGTCTCCGTTGAAGATATTGTTCGTCAGCACTGGGCCACCTATGGTCGCCACTATTACACACGCTATGACTACGAGAACGTTGATGCAGGGGCTGCTAAGGAGCTTATGGCAAACCTAGTCAGCATGCAATCATCGCTTTCTGATGTTAACAAGTTGATCAAGGACATCAGATCTGATGTTTCCGAAGTAGTTGCAGCTGATGAGTTTGAGTACAAGGATCCTGTTGATGGCTCTGTGTCCAAGCACCAGGGTATCCGATACCTCTTTGGAGATGGTTCACGACTGGTGTTCCGTCTCTCTGGAACCGGTTCTGTTGGTGCCACCATCCGTGTCTACATCGAGCAGTACGAGAAGGATTCCTCCAAGACCGGCAGGGATTCACAGGATGCCCTTGCTCCACTGGTTGATGTTGCACTCAAGCTCTCCAAGATGCAAGAGTACACTGGCCGCTCTGCCCCCACCGTGATCACATAA >contig15718 ATGATCCGCCCAGGCTCTAAGGAGTCACAAAATTATGATAATAATAACCAAAAGGTTCATCCTCAACCAATCGATGAGAATATGAATCAGAACGGGGACTCGATGGACACTATGATCGGGAGGATATTCAACAACATATCCTCTTTGAAATCTGCATACATTCAACTGCAAGAAGCCCACACCCCATATGACCCTGACAAGATCCAGGAAGCTGATAAGCTTGTCATAGAGGAGCTTACAAAGCTTTCAGAACTCAAGCATGCTTACAGAGAAAAAAATCCCAAGCCTGTAGCAGCATCCCCTCAAGATTCACGCTTACTTTCTGAAATACAAGAGCAGCAGAATTTGTTGAAGACCTATGAGGTCATGGTAAAGAAGTTCCAGTCCCAGATCCAGACTAGAGATACTGAGATTACCCATTTACAGCAGCAAATAGATGAAGCTAAACTTCGAAAGTCAAAGCTGGAGAAGAAACTGAAACAAAGGGGCCTGCTTAACAAGGAATCAGAGGAATCTGACGAAGAGGAGAACTACTTTTCCATTGAATTGACACCAAGTTTGTTTACATCTGCTGTTGATAACGCATACCAGTCGATGCATGACTTTTCAAAGCCTTTGATCAACATGATGAAGGCTGCAGGTTGGGATCTTGATGCAGCTGCTAATGCAATCGAACCTGCTGTAGTTTACACAAGAAGGGCTCACAAGAAGTATGCTTTTGAATCCTATATTTGCCAAAGAATGTTCAGTGGGTTCCAAGAAGAGAGCTTTTCTATCAAAGATTCTAACATCAGTGTATCCAATGAGGCTTTCTTCCATCAGTTCCTCGCCGTACGAGCCATGGATCCTTTGGATGTCCTCAGCCAAAACCCTGATTCAATTTTTGGCAAGTTCTGCAGAAGCAAATACCTACTACTCGTGCACCCCAAAATGGAAGGTTCTTTCTTCGGTAATATGGATCAGAGGAACTATGTCATGAGCGGTGGCCATCCAAGGACACCTTTCTATCAGGCTTTTCTAAAGCTAGCGAAGTCAATATGGTTGTTGCACAGGCTGGCATACTCCTTTGATCCAAAGGTCAAGGTCTTTCAAGTGAAAAAGGGAAGTGAATTTTCGGATATCCACATGGATAGCGTTGTGAAGAACATCATCCTAGATGAAAGTGCAGAGAGGCTGAAAGTTGGCCTGATGGTGATGCCTGGTTTCTTGATTGGGACCAGCATCATACAGTCCCGGGTGTACCTTTCAGGTATCAAGTGTGCTGACTGA
Functional data
For each type of available functional annotation data (GO terms, protein domains, KO terms), two types of export files are available:
-
Transcripts with functional annotation: a tab-delimited file that contains the transcripts of an experiment and their associated functional annotation labels (identifiers and descriptions). Toggle example -
Functional annotation metadata: a tab-delimited file that contains, for each of every functional annotation label (identifier and description), the number and identifiers of associated transcripts (on a single line). Toggle example
Note: the export of GO functional information has extra columns. The is_hidden column
indicates whether a GO term is flagged as hidden, due to the presence of more informative GO codes in the GO graph for the given transcript,
while the evidence_code column (value set to ISS) indicates that the GO annotation was assigned to the transcript via sequence similarity search.
#counter transcript_id go evidence_code is_hidden description 1 contig00423 GO:0003676 ISS 1 nucleic acid binding 2 contig00423 GO:0003677 ISS 0 DNA binding 3 contig00423 GO:1901363 ISS 1 heterocyclic compound binding 4 contig00423 GO:0097159 ISS 1 organic cyclic compound binding 5 contig00423 GO:0005488 ISS 1 binding 6 contig01755 GO:0003676 ISS 1 nucleic acid binding 7 contig01755 GO:0003677 ISS 0 DNA binding 8 contig01755 GO:0005515 ISS 1 protein binding 9 contig01755 GO:1901363 ISS 1 heterocyclic compound binding 10 contig01755 GO:0097159 ISS 1 organic cyclic compound binding
#counter interpro description num_transcripts transcripts 1 IPR000007 Tubby, C-terminal 17 contig11469 contig05821 contig20328 contig20141 contig15849 contig06374 contig19374 contig19204 contig06372 contig16019 contig06373 contig11470 contig09177 contig20969 contig18970 contig11265 contig18737 2 IPR000008 C2 domain 97 contig08907 contig08909 contig02775 contig07858 contig02773 contig15163 contig02772 contig14957 contig02776 contig22276 contig12203 contig14003 contig05143 contig19878 contig12201 contig09834 contig09835 contig25077 contig04198 contig13022 contig10181 contig04197 contig20905 contig04196 contig16070 contig13823 contig19615 contig14145 contig24751 contig21722 contig18032 contig05835 contig13882 contig14463 contig09510 contig23293 contig09511 contig14358 contig20260 contig14672 contig19081 contig15275 contig19388 contig07086 contig02770 contig02771 contig20071 contig15790 contig04145 contig23085 contig06441 contig04143 contig06440 contig04141 contig11319 contig21842 contig09810 contig09811 contig14049 contig06437 contig06438 contig20391 contig16132 contig18588 contig15968 contig14595 contig14388 contig13965 contig05170 contig13867 contig18699 contig22624 contig23062 contig16332 contig11464 contig11463 contig23722 contig15859 contig23826 contig05678 contig05679 contig18701 contig22117 contig24707 contig05169 contig16583 contig11320 contig23734 contig11321 contig19799 contig21978 contig09893 contig09366 contig04200 contig09368 contig22975 contig14016 3 IPR000009 Protein phosphatase 2A regulatory subunit PR55 7 contig07662 contig03651 contig03650 contig03649 contig07664 contig03647 contig03648 4 IPR000010 Cystatin domain 4 contig21270 contig24646 contig22490 contig21355 5 IPR000011 Ubiquitin/SUMO-activating enzyme E1 5 contig15475 contig15605 contig21279 contig16325 contig14898 6 IPR000014 PAS domain 28 contig24777 contig15287 contig01658 contig15044 contig01654 contig01655 contig16104 contig02922 contig01652 contig01651 contig06672 contig14270 contig06674 contig06675 contig05325 contig05326 contig12570 contig12571 contig12572 contig09082 contig15078 contig05322 contig05323 contig01649 contig15218 contig22643 contig14523 contig13969 7 IPR000023 Phosphofructokinase domain 18 contig15688 contig14681 contig16895 contig15760 contig25248 contig17384 contig15339 contig20162 contig15048 contig10452 contig16255 contig10453 contig10454 contig15002 contig09953 contig09952 contig16755 contig09951 8 IPR000031 PurE domain 1 contig16781 9 IPR000033 LDLR class B repeat 3 contig03829 contig03830 contig03831 10 IPR000039 Ribosomal protein L18e 1 contig19134
Subsets
Subset export files enable the retrieval of the list of sequences that are part of a given transcript subset. They simply consist of a list of sequence identifiers (one identifier per line). Toggle example
contig03137 contig03138 contig03140 contig03141 contig03486 contig03488 contig06873 contig06874 contig08688 contig08689
The toolbox
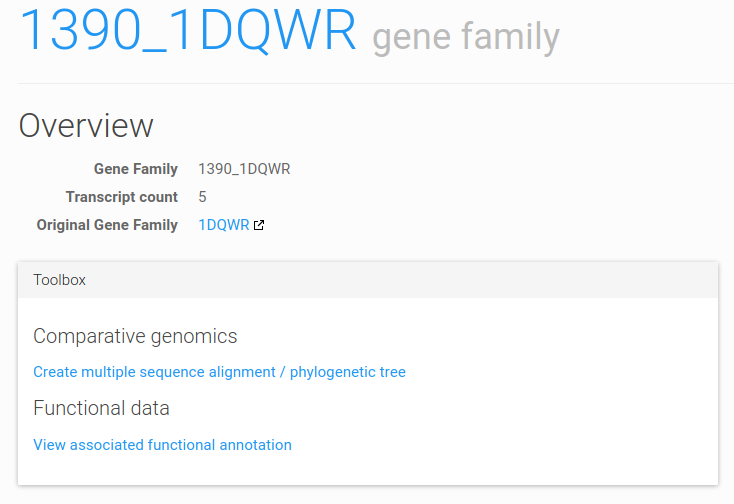
Figure 2: gene family page toolbox. The toolbox is shown on top of any gene family page.
Several pages (e.g. gene family, functional annotation) feature a toolbox that contains links to analyze or visualize the given data object. Figure 2 gives an example of toolbox as shown on the gene family page, which features links to the multiple sequence alignment / phylogenetic tree creation page, and the functional annotation associated to the family.
Analyzing subsets
Apart from the functional annotation of individual transcripts, TRAPID also supports the quantitative analysis of experiment subsets, for instance using functional enrichment statistics (Figure 3).
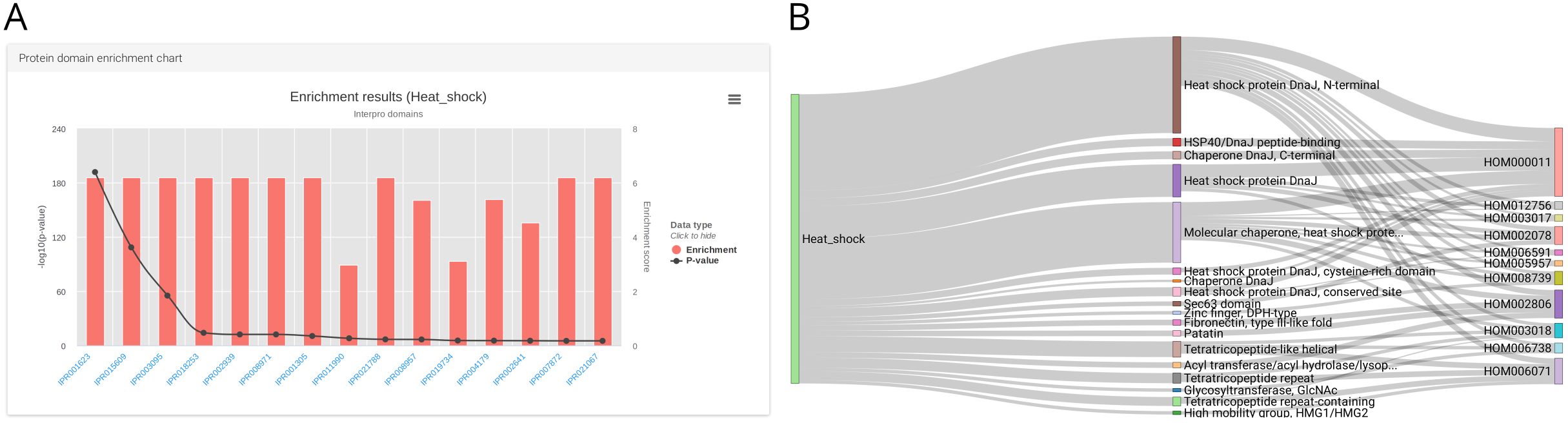
Figure 3: InterPro enrichment results for a subset of MMETSP0936 (Ostreococcus mediterraneus) heat shock transcripts. (A) Enriched InterPro domains visualization. (B) Sankey diagram depicting the relationships between heat shock transcripts (left blocks), significantly enriched InterPro domains (middle blocks), and PLAZA gene families (right blocks).
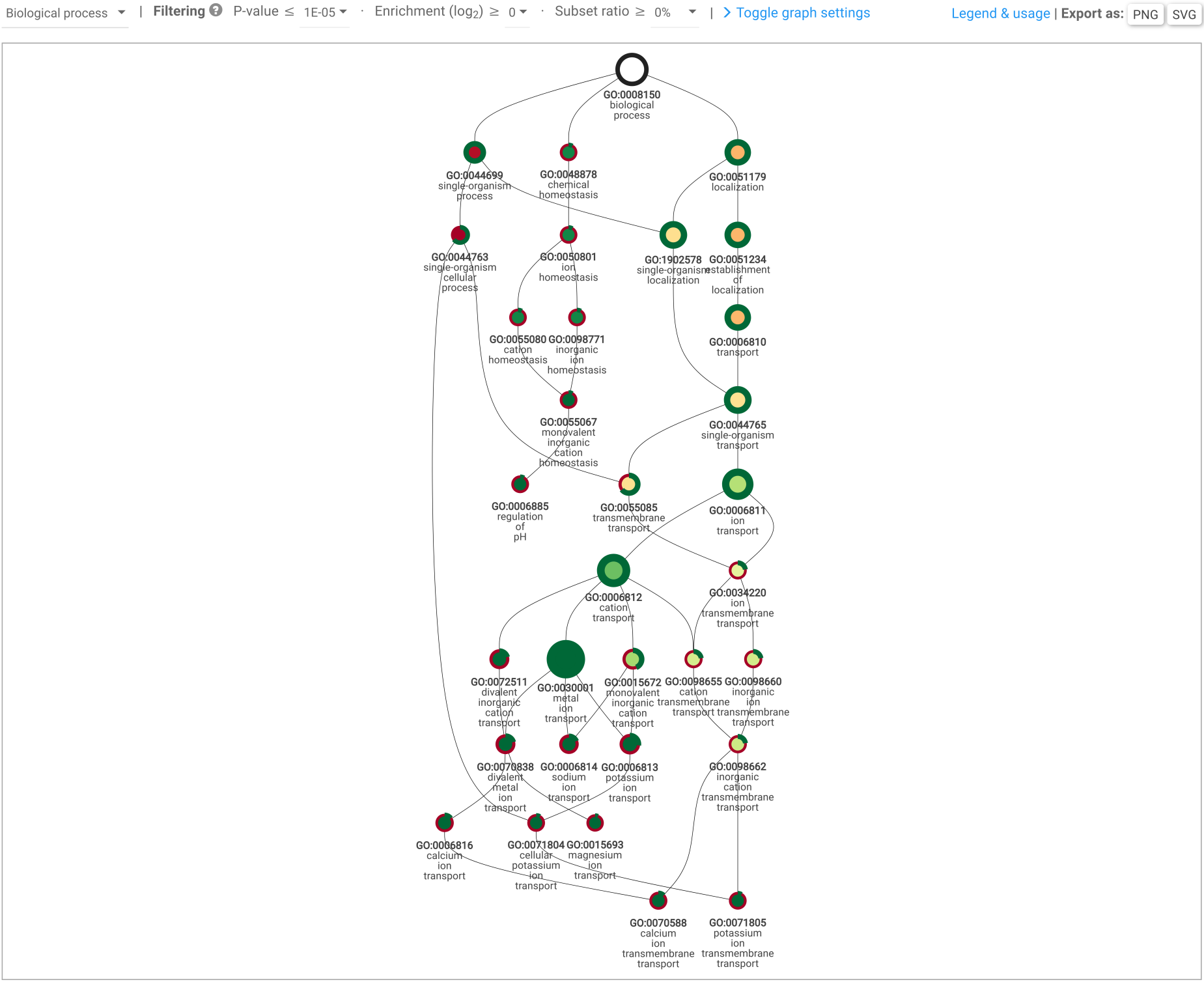
Figure 4: GO enrichment graph example (biological process).
Various comparative analyses can be performed using subsets, accessible from the side menu of an experiment:
Subset enrichment: subset functional enrichment (subset versus all; hypergeometric distribution): visualized as bar chart (Figure 3A), table, Sankey diagrams (Figure 3B), and enrichment GO graph output when working with GO terms (Figure 4).Compare subset: calculates functional annotation frequencies between subsets. Also includes subset-specific annotations.Explore subsets: list subsets, and visualize them as an interactive Venn diagram. From this page, it is also possible to delete subsets.
Taxonomic classification
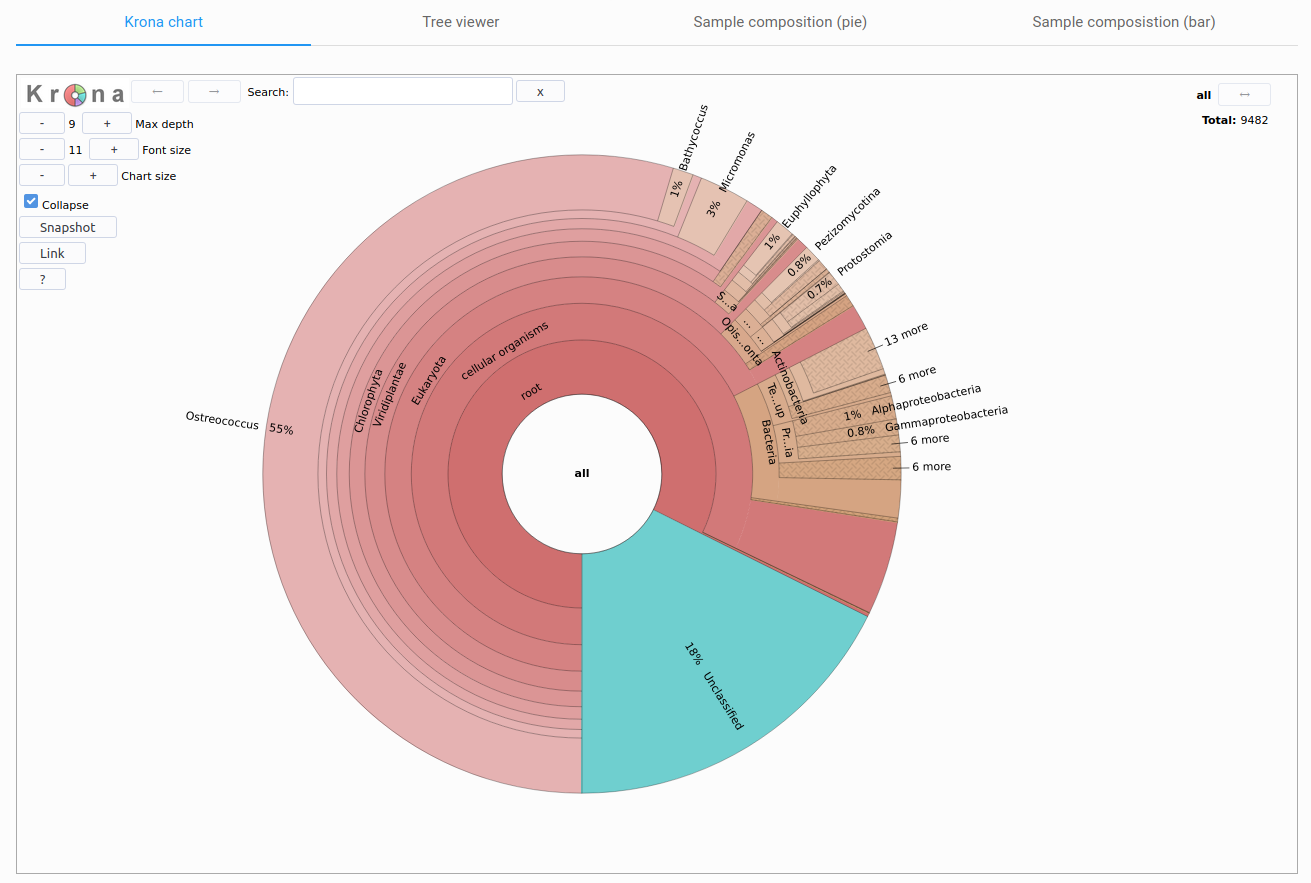
Figure 5: taxonomic classification results visualization. Krona representation of the taxonomic classification results for MMETSP0938 (Ostreococcus mediterraneus).
Taxonomic classification results can be explored by the user through multiple interactive visualizations. To access the taxonomic classification results, click Taxonomic classification in the side menu, within a TRAPID experiment. If this step was not performed during the initial processing, this page is not accessible.
The Krona radial chart, visible in Figure 5, and the tree viewer enable an in-depth examination of the results. In contrast, the sample composition bar and pie charts (Figure 6) provide a quick overview of the results, depicting the domain-level composition and the ten most represented clades at adjustable taxonomic ranks. This page's layout was inspired by EBI Metagenomics.
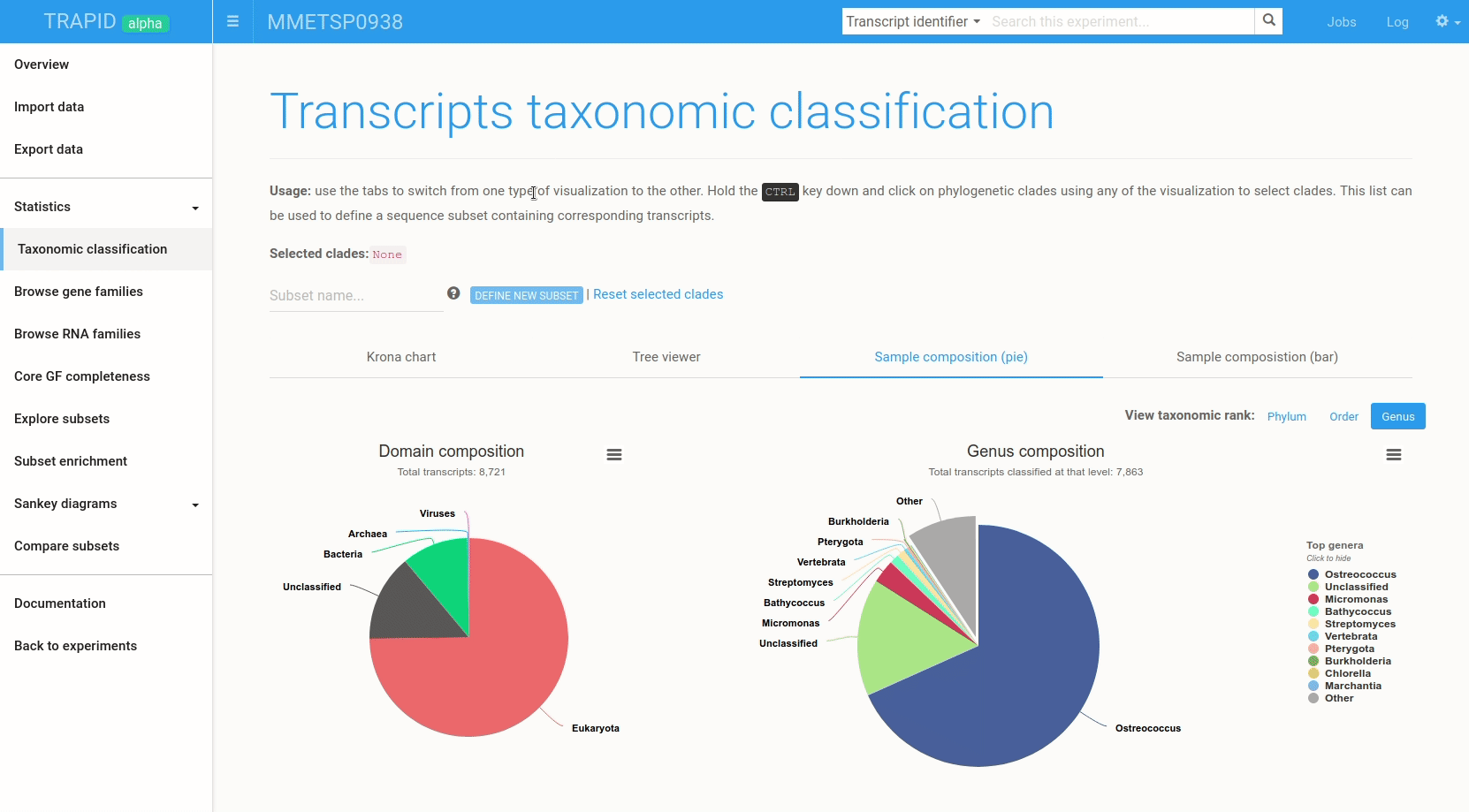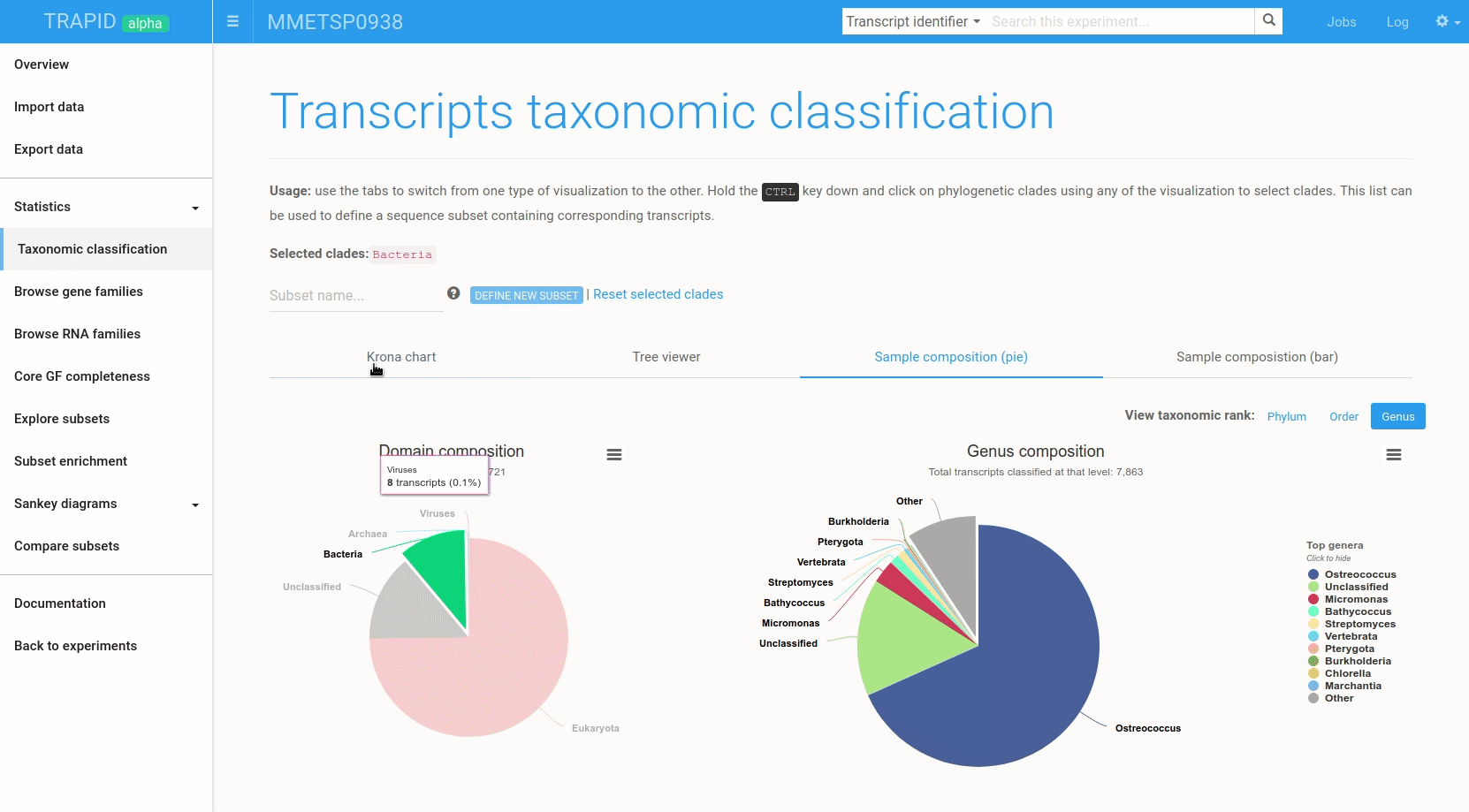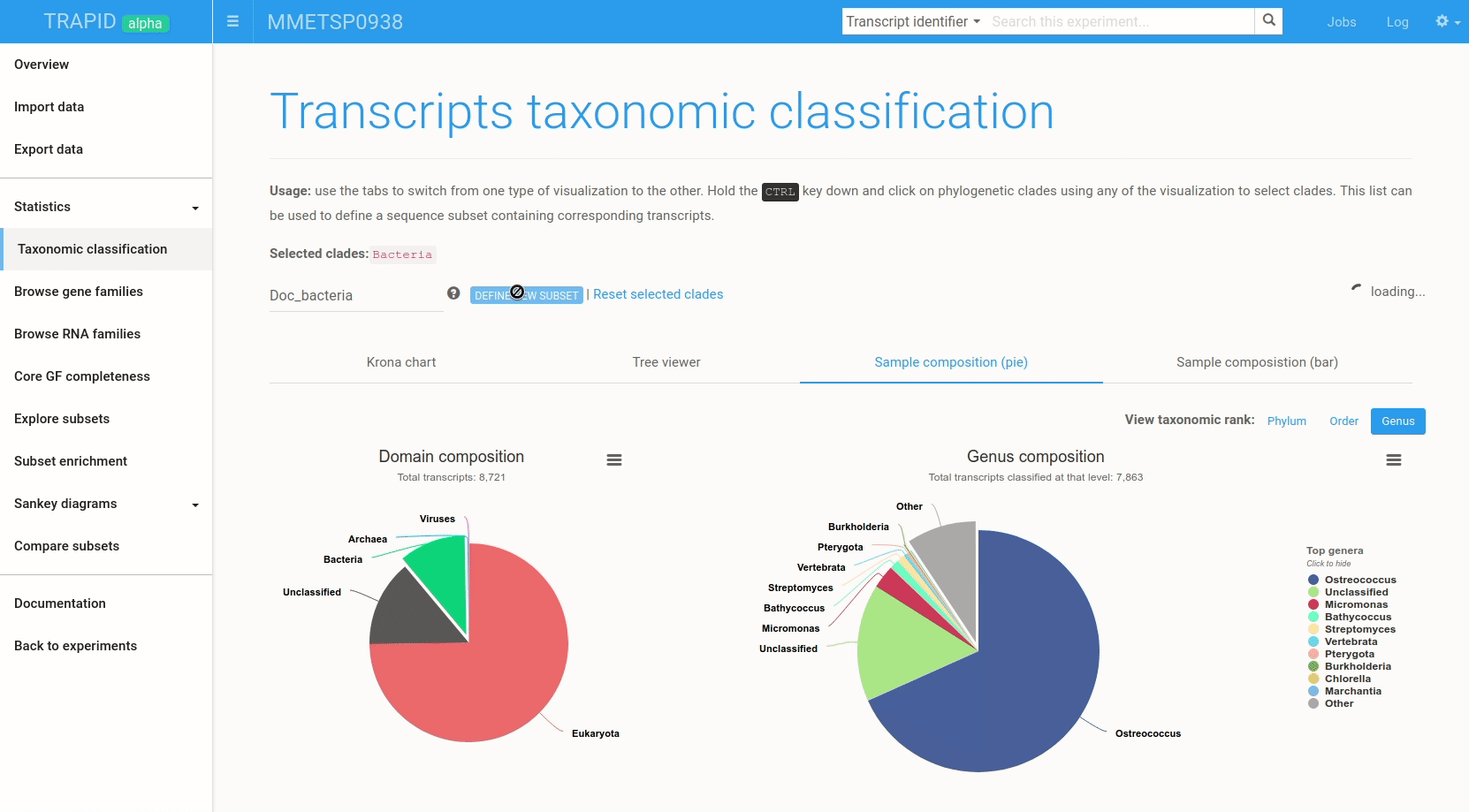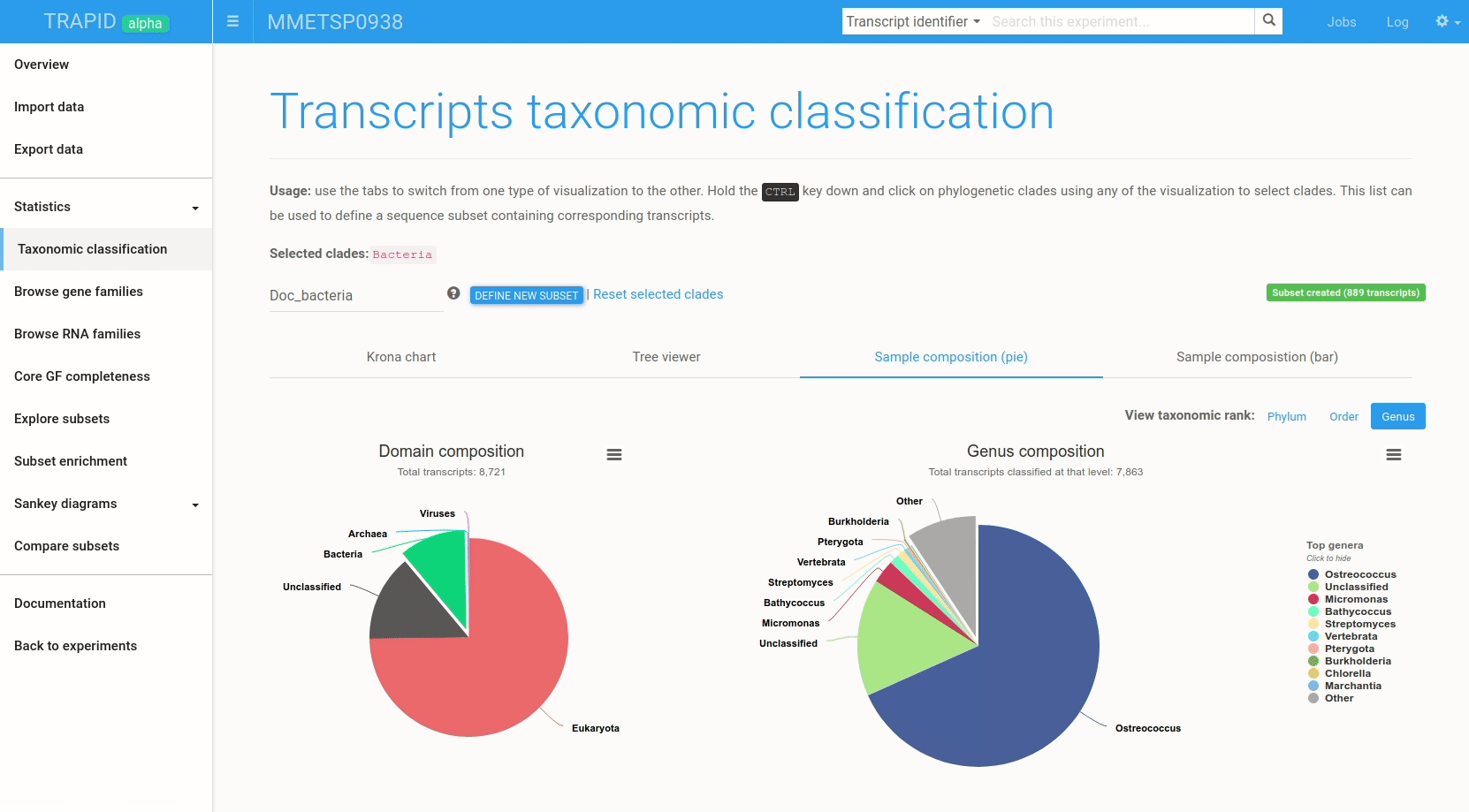
Figure 6: example subset creation from taxonomic classification results. Transcripts assigned to 'bacteria' were selected and grouped in a subset created on-the-fly. The visualized data corresponds to MMETSP0938 (Ostreococcus mediterraneus).
The interactive visualizations can also be used to quickly create transcript subsets by selecting clades: all the transcripts assigned to the selected clade are then grouped in a new transcript subset that can be exported or further analyzed within TRAPID.
To create a subset from the taxonomic classification result page, hold the CTRL key and click on the clades for which a subset should be created. Once the selection is over, type a name for the subset and click create subset. Figure 6 shows an example.
Core GF completeness analysis
TRAPID users can assess and examine the gene space completeness of their input transcriptome data sets, by checking the presence of core gene families (‘core GFs’). Core GFs consist of a set of gene families that are highly conserved in a majority of species within a defined evolutionary lineage. A key feature of this functionality in TRAPID is the on-the-fly definition of core GFs sets for any lineage represented in the selected reference database, making it possible for users to rapidly examine gene space completeness along an evolutionary gradient. The output of the core GF completeness analysis consists of the completeness score (an intuitive quantitative measure of the gene space completeness at the selected taxonomic level ranging between 0-100%), together with the list of represented and missing core GFs and their associated biological functions.
Please note that core gene families are not necessarily single-copy, which is one of the main differences between this approach and the widely-used BUSCO. While the usage of all conserved gene families, regardless of their copy number, enables to cover a much larger fraction of the gene function space, it has nonetheless been shown that core GF completeness and BUSCO scores are generally in agreement (Van Bel et al, 2018)
Step-by-step instructions on how to perform core GF completeness analysis can be found in the tutorial.
Multiple sequence alignments

Figure 7: example multiple sequence alignment. Panicum
data set, transcript contig16311 in family HOM04x5M005933 (PLAZA 4.5 monocots, 52 genes from 35 species).
Starting from a selected transcript, the user has the ability to create an amino acid multiple sequence alignment (MSA) within a gene family context. As such, the user can create an MSA containing the transcripts within a gene family together with a selection of coding sequences from the reference database. This tool is accessible from the toolbox from a gene family (as shown in Figure 2). The MSA is created using MAFFT (default) or MUSCLE. The versions and parameters used for either tool can be found in the tools & parameters page.
After the MSA has been created, the user has the ability to view this
alignment using an in-browser MSA viewer (Figure 5) or to download the MSA and inspect it using a different
tool (Files & extra tab of the MSA/tree page).
Step-by-step instructions on how to generate an MSA can be found in the tutorial.
Phylogenetic trees
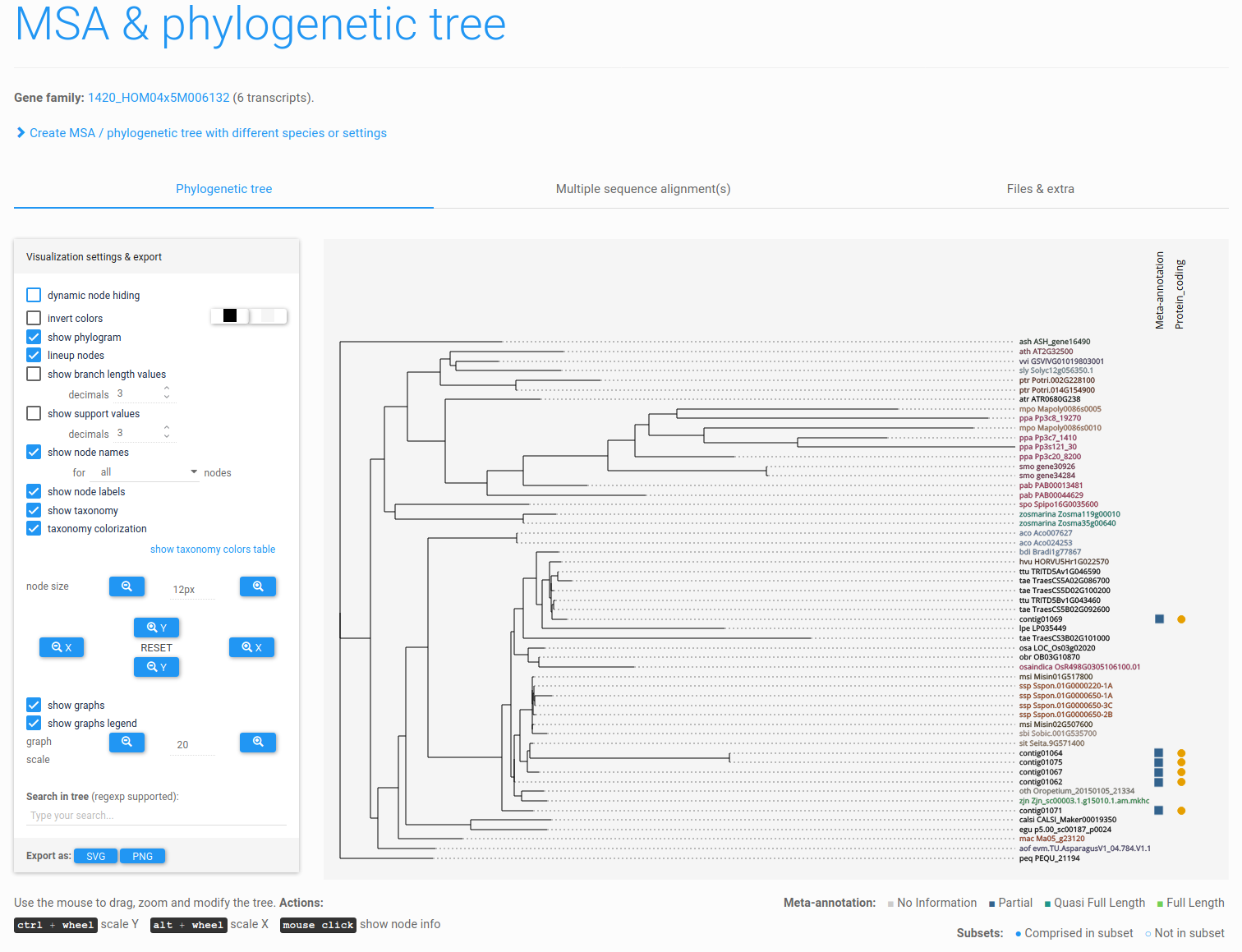
Figure 8: example phylogenetic tree (tutorial). Transcript meta-annotation and subset information are also displayed, depicted next to the transcript identifiers as colored squares and circles, respectively.
Similar to the MSA, the user also has the ability to create a phylogenetic tree within a gene family context (see previous section).
In order to create phylogenetic trees that are less dependent on putative large gaps, the standard MSA can optionally be edited (or stripped). In this stripped MSA the alignment length is reduced by removing input sequences, positions, or both (depending on the user's choice).
In case the editing is too stringent and yields an edited MSA with zero or only a few conserved alignment positions, do not hesitate re-run the tree analysis using a more relaxed editing option (or with no editing altogether, the default), which will yield more conserved alignment positions.
Within TRAPID, four different tree creation programs can be used: FastTree2, IQ-TREE, PhyML, or RaxML. The latter is TRAPID's default due to its very fast processing speed. The versions and parameters used for each tool can be found on the tools & parameters page.
Finally, if the user has defined subsets within his/her experiment, these subsets can also be displayed on the phylogenetic tree, together with meta-annotation, making subsequent analyses much easier. By default, both the subset and meta-annotation information are displayed next to the phylogenetic tree (see Figure 8).
Step-by-step instructions on how to create phylogenetic trees can be found in the tutorial.
Orthology
A key challenge in comparative genomics is reliably grouping homologous genes (derived from a common ancestor) and orthologous genes (homologs separated by a speciation event) into gene families. Orthology is generally considered a good proxy to identify genes performing a similar function in different species. Consequently, orthologs are frequently used as a means to transfer functional information from well-studied model systems to non-model organisms, for which e.g. only RNA-Seq-based gene catalogs are available. In eukaryotes, the utilization of orthology is not trivial, due to a wealth of paralogs (homologous genes created through a duplication event) in almost all lineages. Ancient duplication events preceding speciation led to outparalogs, which are frequently considered as subtypes within large gene families. In contrast to these are inparalogs, genes that originated through duplication events occurring after a speciation event. Besides continuous duplication events (for instance, via tandem duplication), many paralogs are remnants of different whole genome duplications (WGDs), resulting in the establishment of one-to-many and many-to-many orthologs (or co-orthologs).
Within TRAPID, the phylogenetic trees provide the most detailed approach to identify orthology relationships. For a given transcript, inspecting the phylogenetic tree can reveal whether orthologs exist in related species and if this relationship is a one-to-one, one-to-many, or many-to-many orthology. Apart from the trees, also the Browse similarity search output tool, available from a transcript page, offers an overview of homologous genes in related species. Below, we show two examples demonstrating how orthologous groups and simple/complex orthology relationships can be derived from a phylogenetic tree generated using TRAPID.
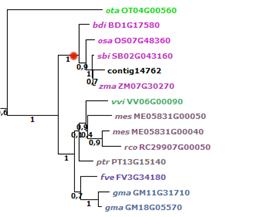
Figure 9: example one-to-one orthology
(Panicum data set, transcript contig14762 RecQ helicase). The node indicated in red
shows the monocot homologs for this clade (1 indicates 100% bootstrap support). Within this
sub-tree, from each included monocot species a single gene is present, revealing simple
one-to-one orthology relationships.
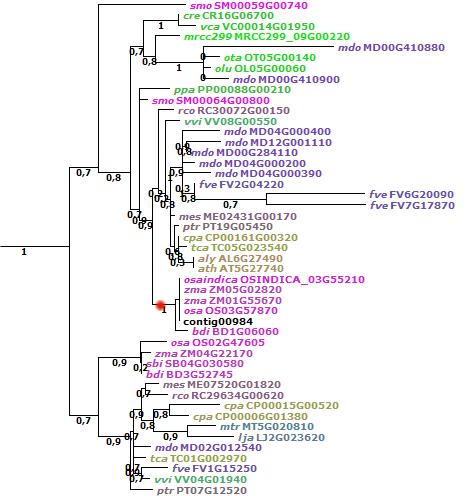
Figure 10: example one-to-many orthology
(Panicum data set, transcript contig00984 ATPase). The node indicated in red shows
the monocot homologs for this clade (1 indicates 100% bootstrap support). Within this
sub-tree, 2 genes from Zea mays are present, revealing that for the single Panicum
transcript two co-orthologs exist in Z. mays.
Appendix
Sequence assembly resources
Multiple online resources provide researchers with computational infrastructure and graphical user-interfaces to use state-of-the-art sequence assembly tools (e.g. rnaSPAdes or Trinity for de novo transcriptome assembly) and prepare their input data for use with TRAPID.
For example:
-
Galaxy is a web-based solution that provides computational resources and graphical interfaces to facilitate the use of bioinformatics software.
- Both the main and European Galaxy instances feature multiple tools for sequence assembly.
- Numerous other Galaxy instances are publicly available: please browse the Galaxy platform directory for more information.
- The CyVerse cyberinfrastructure also provides a web-based analytical platform on which it is possible to run various sequence assembly tools.
TRAPID processing steps and their purpose
The below table provides an overview of all TRAPID processing steps and their purpose. Processing steps are organized by processing phase — initial processing or exploratory — and execution order when possible.
| Processing phase | Processing step | Purpose |
|---|---|---|
| Initial processing phase | Global transcriptome characterization | |
| Taxonomic classification | Potential contaminant detection; characterization of communities | |
| RNA transcripts detection | Non-coding RNA identification and annotation | |
| Sequence similarity search | Identification of homologous sequences within available reference databases | |
| Sequence similarity search post-processing | Characterization of protein-coding transcripts | |
| — Gene Family assignment | Clustering of transcripts with their homologs | |
| — Functional annotation transfer | Functional annotation of transcript sequences | |
| — Meta-annotation inference | Estimation of the fragmentation state of transcripts | |
| — Frame detection and ORF finding | Putative frameshift detection; coding sequence detection and translation (supporting non-canonical genetic codes) | |
| Exploratory phase | Refined comparative and functional analyses; examination of selected data types | |
| Experiment statistics | Exploration of annotation metrics and sequence length distribution | |
| Global protein alignment and phylogenetic tree construction | Analysis of transcripts and their homologs in a gene-family and evolutionary context; identification of orthology and paralogy relationships | |
| Core GF completeness analysis | Gene space completeness assessment and analysis at multiple phylogenetic levels | |
| Subset functional enrichment analysis | Identification and analysis of functional biases; discovery and exploration of significantly over-represented biological functions | |
| Subset functional annotation comparison | Exploration of the biological function of transcript subsets; comparison of functional biases |