Workbench
The workbench is a tool to analyze multiple genes in batch through user-defined gene sets. When manually browsing genes on a one-by-one basis is no longer feasible, you can store different sets of genes in your workbench and analyze them using PLAZA's toolbox.
This tutorial will explain how to import/export gene sets in the workbench and how to analyze them using the different tools.
Creating an account

Your workbench is private and is protected using your e-mail address and a password. To prevent unauthorized users from accessing your data a log-in system is installed (Figure 1). If you don't have an account yet, you need to create one (Analyze → Workbench). Provide a valid e-mail address and an auto-generated password will be sent (can be changed afterward). Once you received your password, return to this page to log in.
Adding genes to your workbench
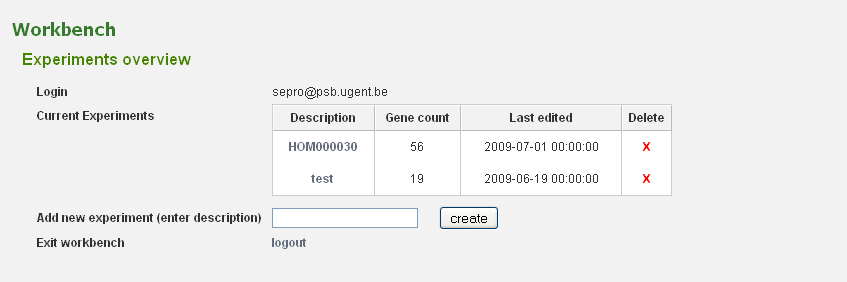
Before genes can be added a new experiment has to be created. Once logged in with a valid e-mail address and password, an overview of the contents of your workbench is provided (can be reached through Analyze → Workbench at any time). Fill in an appropriate name for your experiment and hit the create button. (Figure 2) Next select your experiment from the list. This will take you to a particular experiment. The last thing you need to do before you can perform an analysis is adding a set of genes to your selected experiment. Currently three methods are available to import genes to your experiment: import using gene lists, sequence similarity search or import using a BED file. (Figure 3)
- Add Genes: in your experiment overview click import genes and in the textbox fill in a list of genes you would like to add. You can use PLAZA gene identifiers or external identifiers provided by the original annotation provider(s). In the latter case a conversion table will be displayed with the internal PLAZA gene id.
- BLAST-search: using a FASTA sequence file (with e.g. EST data) a best BLAST-hit procedure against PLAZA genes from a selected reference organism can be applied to import genes in the workbench. This way sequence data from a non-model organism can be mapped to an organism included in PLAZA. You will receive an e-mail when the BLAST-based gene import is ready. Note that the link between the original accession number of your sequence and the PLAZA gene identifier is specified in the comment field (together with the BLAST E-value). When multiple sequences map to the same reference gene no redundant entries are created.
- BED import: a Browser Exstensible Data (BED) file, containing genomic locations for specific features, can be used to import genes into the workbench. The PLAZA workbench uses BEDOPS to map the regions to the genes of a selected reference organism, and imports these obtained genes into the workbench experiment. The mapping is dependent on a given maximum distance metric and, if more than one gene lies within that distance, then only the closest gene is selected. If the same gene is associated with more than one region, it will be included only once and with the absolute shortest distance.
- In the experiment overview unwanted genes can be removed as well by clicking the X.
A step-by-step example
In this tutorial the workbench will be used to identify in which biological processes auxin-related genes are involved. In addition, by mapping all genes to the associated gene families an evolutionary analysis can be performed using all genes simultaneously.
Finding the genes and loading them in the workbench

- To start we need to collect all gene identifiers of genes related to auxin. This will be done using a keyword search on gene descriptions: in the Search bar type auxin, change all species to Arabidopsis thaliana and hit search.
- This query returns a list with 287 genes that have the word auxin in their description. At the bottom of the results page in the Download genes section, de-select everything except gene_id in 'include following columns' and click generate result file.
- Save the file to your local hard disk and open it with a text editor (like WordPad, SciTE, Emacs, etc.)
- Remove the first line: #gene_id
- Now go to your workbench (Analyze→Workbench) and log in. Create a new experiment and add all the genes from the text file to this experiment. If you are not familiar with how to create an experiment and add genes, please check the previous section of this tutorial (here)
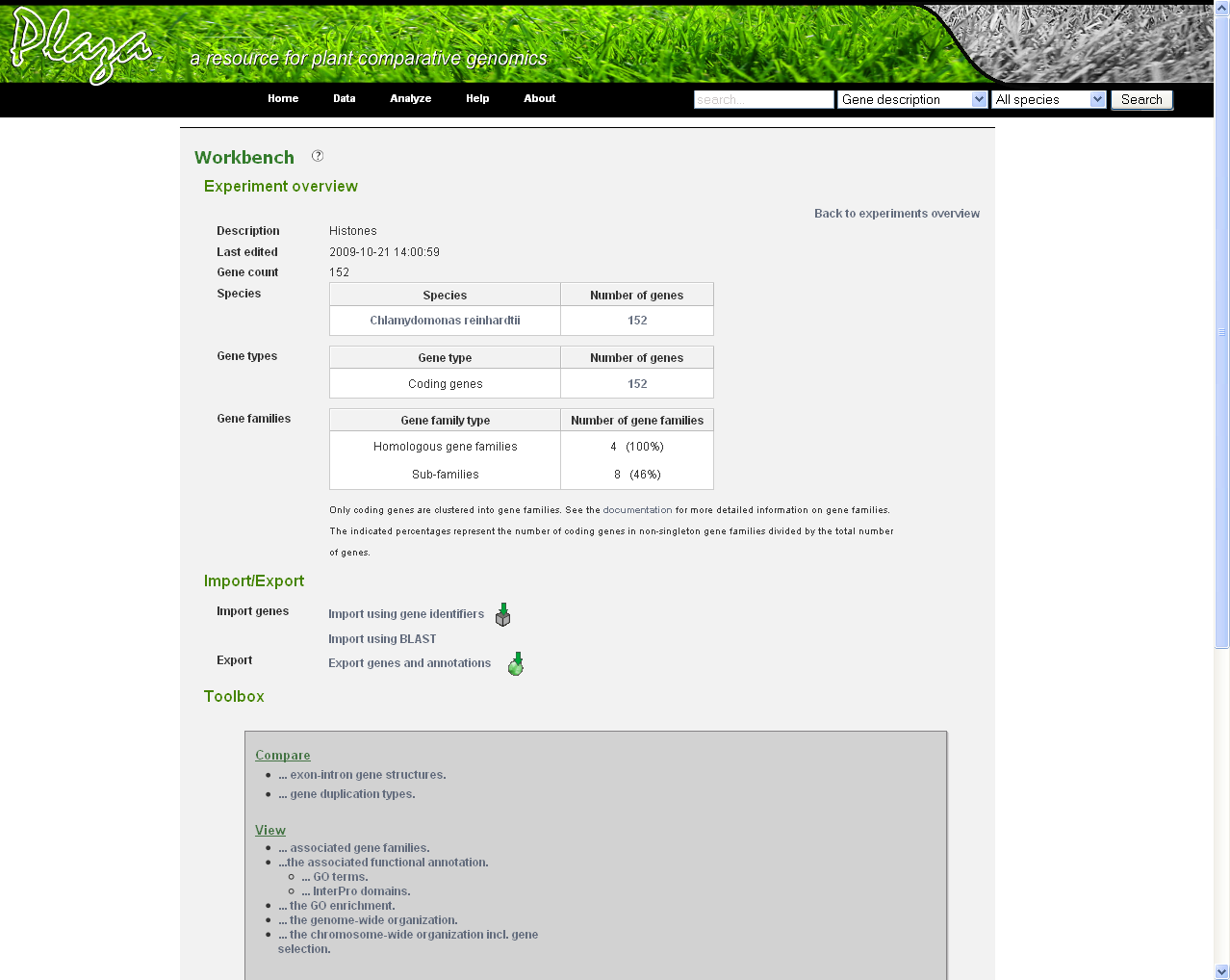
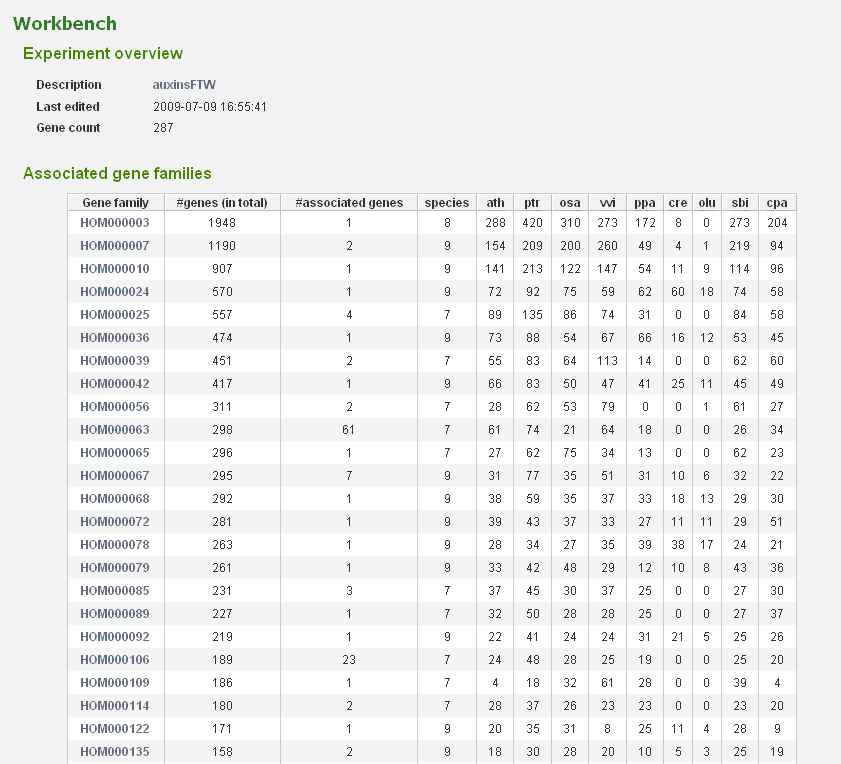
- In the Workbench overview (Analyze→Workbench), click on the newly created experiment. On this page some statistics are shown about the genes included in the dataset.
Using the workbench
- On this page there is a toolbox providing links to different features:
- To compare the exon-intron structure of the included genes, click Compare exon-intron gene structures. Note that all genes are sorted based on the gene family they belong to. (Figure 4)
- To get more information about the duplication types (i.e. presence of block or tandem duplicates) of the genes in the dataset, go to Compare gene duplication types

- The associated gene families can be inspected to find homologous genes in other species or to find paralogs that might lack a proper description and are therefore missed in the keyword search. Through the phylogenetic trees detailed information about orthologs and paralogs can be obtained (per gene family). (Figure 5)
- When focussing on gene families with multiple (auxin-)associated genes, we note that several of them do not have homologs in Chlamydomonas and Ostreococcus, suggesting that this hormone is not present in green algae.
- Complementary, clicking on gene family HOM000063 covering auxin-responsive proteins reveals that multiple sub-families can be found, indicating massive duplication-mediated gene family expansion. In addition, copy numbers are also higher in eudicots compared to monocots or moss. Inspecting the similarity heatmap or the phylogenetic trees for the sub-family of gene AT2G21200 (ORTHO000231) indeed reveals numerous species-specific duplicates in several eudicots.
- To display more details about the functional annotation of the selected genes, click View GO terms or View InterPro domains
- The distribution of the genes on the different chromosomes can be inspected using View the genome wide organization (Figure 6)
- The last feature is the GO enrichment tool. Clicking View the GO enrichment in the toolbox will guide you to the enrichment analysis page. Based on your selected genes GO terms are collected and bar charts are shown with all significantly enriched/depleted functional categories in the experiment. By default some filters are applied to hide redundant GO terms (caused by parent-child relations) in the charts.
- On the bottom a full overview of all enriched/depleted GO-labels is shown with enrichment folds and p-values. GO terms tagged with a green V are plotted in the charts.
- In the options box on the right set the cut-off value to 0.01.
Interpretation of the results
Now let's inspect the output! Remember we started with a set of genes related to auxin to figure out which processes this hormone is involved in. So we'll go over a few of the enriched GO labels in the category Biological process:
- A first set is related with root development and lateral root initiation/development
- GO:0022622: root system development, GO:0048364: root development, GO:0048527: lateral root development, GO:0010102: lateral root morphogenesis, GO:0010311 lateral root formation, GO:0048830: adventitious root development
- Also shoot, leaf and flower development seem to be closely associated with auxins.
- GO:0022621: shoot system development, GO:0048827: phyllome development, GO:0048366: leaf development, GO:0048438: floral whorl development, GO:0010016: shoot morphogenesis
- There are several responses to the environment enriched as well
- GO:0009629: response to gravity, GO:0009630: gravitropism, GO:0009958:positive gravitropism, GO:0009640:photomorphogenesis, GO:0009639: response to red or far red light, GO:0002237: response to molecule of bacterial origin, GO:0009641:shade avoidance
- A link between auxin and other plant hormones can be observed
- GO:0009735:response to cytokinin stimulus, GO:0009723:response to ethylene stimulus
Conclusion
So actually this analysis is able to identify several auxin related processes. Root and shoot development are the textbook examples: a difference in auxin concentration can effect the growth of the plant asymmetrically to cause tropism. More surprising is that a link between auxin, cytokinin and ethylene was found: the ratio of auxin/cytokinin is known to cause either root or xylem initiation in callus. An excess of auxins can block ethylene.