Contents
?
Installing
ForCon
After downloading ForCon, you have a .zip file. Unzip it using WinZip
or PKUNZIP in a temporary directory. Double-click the setup.exe file and
follow the instructions.
?
General
description
ForCon is a user-friendly software tool for the easy conversion of nucleic
and amino acid sequence alignments into different formats.
At the moment, ForCon is able to convert ý in both ways, i.e. reading
and writing - the
following formats (or formats used by the following software packages):
?
-
CLUSTAL
-
EMBL
-
FASTA
-
GCG/MSF
-
Hennig86
-
MEGA
-
NBRF/PIR
-
PAUP/Nexus
-
Parsimony Jackknifer
-
PHYLIP
-
TREECON
Software packages not included in the list are usually able to read
one of the formats mentioned.?? For the publication of sequence
alignments, a format with codon positions can be generated ("Pretty").
Sequential and interleaved formats are supported by ForCon. (see next
paragraph)
?
?
File
formats
The use of correct formats is extremely important: incorrect formats
cannot be correctly interpreted by the program. For this reason a description
and example of all the formats is presented below.
Overall, two major types of formats exist: interleaved and noninterleaved
(sequential). In the interleaved format, sequences are written in the form
of an alignment:
?Sequence 1?? AGUCGAGUC---GCAGAAACGCAUGAC
?Sequence 2?? AGUCGCGUCG--GCAGAAACGCAUGAC
?Sequence 3?? AGUCGCGUCG--GCAGAUACGCAUCAC
?Sequence 4?? AGUCGCGUCGAAGCAGA--CGCAUGAC
(Sequence 1)? -GACCACAUUUU-CCUUGCAAAG
(Sequence 2)? GGACCACAUCAU-CCUUGCAAAG
(Sequence 3)? GGAC-ACAUCAUCCCUCGCAGAG
(Sequence 4)? GGACCACAUCAUCCCUUGCAGAG
In the noninterleaved formats, sequences are written one after another:
Sequence 1??????
AGUCGAGUC---GCAGAAACGCAUGAC
-GACCACAUUUU-CCUUGCAAAG
Sequence 2?? AGUCGCGUCG--GCAGAAACGCAUGAC
GGACCACAUCAU-CCUUGCAAAG
Sequence 3??? AGUCGCGUCG--GCAGAUACGCAUCAC
GGAC-ACAUCAUCCCUCGCAGAG
Sequence 4?????
AGUCGCGUCGAAGCAGA--CGCAUGAC
GGACCACAUCAUCCCUUGCAGAG
Usually the symbol for missing data is 'N' (nucleotides) or 'X' (proteins).
For insertions/deletions ('gaps') the most commonly used symbol is a hyphen
'-'.
Regarding the different formats:
?
1) CLUSTAL
The CLUSTAL program is a program for creating sequence alignments.
The CLUSTAL format can be described as follows:
- the word CLUSTAL should be on the first non-space line of the file
- the alignment is displayed in blocks of a fixed length
- each line in the block corresponds to one sequence
- the line starts with the sequence name (of any length), followed
by at least one space character
- then the sequence itself is displayed (upper- or lowercase) ( '-'
: gaps )
??? (optional : residue number at the end)
- in between blocks: line with conservation info ( ForCon only writes
stars for now ; for more info:
??? http://www-igbmc.u-strasbg.fr/BioInfo/ClustalX/#G
)
Example :
CLUSTAL W (1.74) multiple sequence alignment
Homo_sapiens???????
AGUCGAGUC---GCAGAAAC
Pan_paniscus???????
AGUCGCGUCG--GCAGAAAC
Gorilla_gorilla???? AGUCGCGUCG--GCAGAUAC
Pongo_pigmaeus?????
AGUCGCGUCGAAGCAGA--C
???????????????????
***** ***?? *****? *
Homo_sapiens???????
GCAUGAC-GACCACAUUUU-
Pan_paniscus???????
GCAUGACGGACCACAUCAU-
Gorilla_gorilla???? GCAUCACGGAC-ACAUCAUC
Pongo_pigmaeus?????
GCAUGACGGACCACAUCAUC
???????????????????
**** ** *** ****? *
Homo_sapiens???????
CCUUGCAAAG
Pan_paniscus???????
CCUUGCAAAG
Gorilla_gorilla???? CCUCGCAGAG
Pongo_pigmaeus?????
CCUUGCAGAG
???????????????????
*** *** **
2) EMBL
The EMBL database is the primary nucleotide database in Europe.
The format is described in detail at: http://www.ebi.ac.uk/ebi_docs/embl_db/usrman/structure_entry.html
Multiple sequence files also follow these rules. They are separated
by the '//' that ends each entry.
Only the information used in multiple sequence alignments is used by
ForCon.
Example ( as generated by ForCon; for input, any EMBL file is allowed
):
ID?? Homo sapiens
SQ?? Sequence 50 BP;
???? AGUCGAGUC- --GCAGAAAC
GCAUGAC-GA CCACAUUUU- CCUUGCAAAG
//
ID?? Pan paniscus
SQ?? Sequence 50 BP;
???? AGUCGCGUCG --GCAGAAAC
GCAUGACGGA CCACAUCAU- CCUUGCAAAG
//
ID?? Gorilla gorilla
SQ?? Sequence 50 BP;
???? AGUCGCGUCG --GCAGAUAC
GCAUCACGGA C-ACAUCAUC CCUCGCAGAG
//
ID?? Pongo pigmaeus
SQ?? Sequence 50 BP;
???? AGUCGCGUCG AAGCAGA--C
GCAUGACGGA CCACAUCAUC CCUUGCAGAG
//
3) FASTA
The FASTA program is used for database searches.
The format is described at : http://www.ncbi.nlm.nih.gov/BLAST/fasta.html
Example:
>Homo sapiens
AGUCGAGUC---GCAGAAACGCAUGAC-GACCACAUUUU-CCUUGCAAAG
>Pan paniscus
AGUCGCGUCG--GCAGAAACGCAUGACGGACCACAUCAU-CCUUGCAAAG
>Gorilla gorilla
AGUCGCGUCG--GCAGAUACGCAUCACGGAC-ACAUCAUCCCUCGCAGAG
>Pongo pigmaeus
AGUCGCGUCGAAGCAGA--CGCAUGACGGACCACAUCAUCCCUUGCAGAG
?
4) GCG/MSF
The Multiple Sequence File format by the Genetics Computer Group Wisconsin
package is thoroughly described in their user manual. In brief:
- on the first line : file type identifier like '!!AA_MULTIPLE_ALIGNMENT
1.0',
??? '!!NA_MULTIPLE_ALIGNMENT 1.0' or 'PileUp'. ( optional
)
- second line: optional title/description
- dividing line with obligatory 'MSF: sequence length', checksum value
and two points '..'
- name/weight section with checksum
- separating line : //
- alignment : interleaved
Example ( as generated by ForCon? )
!!NA_MULTIPLE_ALIGNMENT 1.0
Four anthropoidea
MSF: 50? Type: N? Check: 2666 ..
Name: Homo_sapiens????
Len: 50?? Check: 8318?? Weight: 1.00
Name: Pan_paniscus????
Len: 50?? Check: 7854?? Weight: 1.00
Name: Gorilla_gorilla? Len: 50??
Check: 7778?? Weight: 1.00
Name: Pongo_pigmaeus?? Len: 50??
Check: 8716?? Weight: 1.00
//
Homo_sapiens???????
AGUCGAGUC...GCAGAAAC
Pan_paniscus???????
AGUCGCGUCG..GCAGAAAC
Gorilla_gorilla???? AGUCGCGUCG..GCAGAUAC
Pongo_pigmaeus?????
AGUCGCGUCGAAGCAGA..C
Homo_sapiens???????
GCAUGAC.GACCACAUUUU.
Pan_paniscus???????
GCAUGACGGACCACAUCAU.
Gorilla_gorilla???? GCAUCACGGAC.ACAUCAUC
Pongo_pigmaeus?????
GCAUGACGGACCACAUCAUC
Homo_sapiens???????
CCUUGCAAAG
Pan_paniscus???????
CCUUGCAAAG
Gorilla_gorilla???? CCUCGCAGAG
Pongo_pigmaeus?????
CCUUGCAGAG
?
?
5) Hennig86
The parsimony phylogeny program by Farris uses an unusual format: the
different IUPAC nucleotide letter codes are replaced by a number code.
ForCon uses the following standard translation :
?
?
|
A
|
to:
|
0
|
|
U,T
|
to:
|
1
|
|
G
|
to:
|
2
|
|
C
|
to:
|
3
|
|
N
|
to:
|
?
|
When converting from the Hennig86 format, the user will be prompted
to enter his/her own translation preferences.
The format is a sequential format. On the first line there is the word
'xread', used for recognition of the file. On the following line a title/description
can be placed in between single quotes. The third line consists of the
sequence length and the number of sequences. After the alignment ( is sequential
format ), the file is closed by a semicolon (;). The symbol used for missing
data is '?'. There is no separate character for defining gaps.
Example:
xread
' Four anthropoidea '
50 4
Homo sapiens
132431324???341311143412314?31441412222?4422341113
Pan paniscus
1324343243??341311143412314331441412412?4422341113
Gorilla gorilla
1324343243??3413121434124143314?141241244424341313
Pongo pigmaeus
13243432431134131??4341231433144141241244422341313
;
6) MEGA
The Molecular Evolutionary Genetic Analysis program by Kumar, Tamura
& Nei is a tree construction program based on distance- and parsimony
methods.
The format? is described in the MEGA manual. In brief:
The format exists in the interleaved and noninterleaved format.
Disregarding the format type, the file always starts with the word
'#mega' on the first line. On the following line, a title can be stated,
preceded by the term 'TITLE:'. In between the title and the sequence data,
a description or extra comments can be placed. Even inside the sequences,
comments are allowed in between quotes (""). The sequence names are preceded
by a '#'.
Examples:
#mega
TITLE: Four Anthropoidea
The interleaved format
#Homo_sapiens???????
AGUCGAGUC---GCAGAAACGCAUGAC-GACC
#Pan_paniscus???????
AGUCGCGUCG--GCAGAAACGCAUGACGGACC
#Gorilla_gorilla????
AGUCGCGUCG--GCAGAUACGCAUCACGGAC-
#Pongo_pigmaeus?????
AGUCGCGUCGAAGCAGA--CGCAUGACGGACC
#Homo_sapiens???????
ACAUUUU-CCUUGCAAAG
#Pan_paniscus???????
ACAUCAU-CCUUGCAAAG
#Gorilla_gorilla????
ACAUCAUCCCUCGCAGAG
#Pongo_pigmaeus?????
ACAUCAUCCCUUGCAGAG
---
#mega
TITLE: Four Anthropoidea
The noninterleaved format
#Homo_sapiens
AGUCGAGUC---GCAGAAACGCAUGAC-GACCACAUUUU-CCUUGCAAAG
#Pan_paniscus
AGUCGCGUCG--GCAGAAACGCAUGACGGACCACAUCAU-CCUUGCAAAG
#Gorilla_gorilla
AGUCGCGUCG--GCAGAUACGCAUCACGGAC-ACAUCAUCCCUCGCAGAG
#Pongo_pigmaeus
AGUCGCGUCGAAGCAGA--CGCAUGACGGACCACAUCAUCCCUUGCAGAG
?
7) NBRF/PIR
The format of this large protein database is similar
to the FASTA format. Each sequence, though, starts with a '>[sequence type
code];', followed by the sequence name and a description ( on the next
line ).
This description is ignored by ForCon.
On the following line the actual sequence is
written and is ended with an asterisk (*).
The sequence type codes are as follows:
?
?
|
Code
|
Sequence type
|
| P1 |
Protein (complete) |
|
F1
|
Protein (fragment)
|
|
DL
|
DNA (linear)
|
|
DC
|
DNA (circular)
|
|
RL
|
RNA (linear)
|
|
RC
|
RNA (circular)
|
|
N3
|
tRNA
|
|
N1
|
other functional RNA
|
ForCon accepts all these codes, but only writes down codes P1, D1 and
RL.
Example :
>RL;Homo sapiens
Homo sapiens RNA sequence
AGUCGAGUC---GCAGAAACGCAUGAC-GACCACAUUUU-CCUUGCAAAG*
>RL;Pan paniscus
Pan paniscus RNA sequence
AGUCGCGUCG--GCAGAAACGCAUGACGGACCACAUCAU-CCUUGCAAAG*
>RL;Gorilla gorilla
Gorilla gorilla RNA sequence
AGUCGCGUCG--GCAGAUACGCAUCACGGAC-ACAUCAUCCCUCGCAGAG*
>RL;Pongo pigmaeus
Pongo pigmaeus RNA sequence
AGUCGCGUCGAAGCAGA--CGCAUGACGGACCACAUCAUCCCUUGCAGAG*
?
8) PAUP/NEXUS
The Nexus format is used by several programs: PAUP, MacClade, Spectrum,...
.
For a detailed description of the format, I'd like to refer to the
article written by Maddison et al. :
Maddison, D.R., Swofford, D.L., Maddison, W.P.
(1997) NEXUS: An extendible file format for systematic information. Syst.Biol.
46, 590-621.
ForCon is limited in the use of this extremely versatile format. Only
the information on the alignment itself is used and generated, although
any NEXUS file can be used as input file. The program will ignore all information
that is not used.
Here is an example of a NEXUS file generated by the ForCon program:
#NEXUS
[TITLE: Four Anthropoidea]
begin data;
dimensions ntax=4 nchar=50;
format datatype=RNA missing=N gap=-;
matrix
Homo_sapiens
AGUCGAGUC---GCAGAAACGCAUGAC-GACCACAUUUU-CCUUGCAAAG
Pan_paniscus
AGUCGCGUCG--GCAGAAACGCAUGACGGACCACAUCAU-CCUUGCAAAG
Gorilla_gorilla
AGUCGCGUCG--GCAGAUACGCAUCACGGAC-ACAUCAUCCCUCGCAGAG
Pongo_pigmaeus
AGUCGCGUCGAAGCAGA--CGCAUGACGGACCACAUCAUCCCUUGCAGAG
;
endblock;
begin assumptions;
options deftype=unord;
?
---
?
#NEXUS
[TITLE: Four Anthropoidea]
begin data;
dimensions ntax=4 nchar=50;
format interleave datatype=RNA missing=N
gap=-;
matrix
Homo_sapiens???????
AGUCGAGUC---GCAGAAACGCAUGAC-GAC
Pan_paniscus???????
AGUCGCGUCG--GCAGAAACGCAUGACGGAC
Gorilla_gorilla???? AGUCGCGUCG--GCAGAUACGCAUCACGGAC
Pongo_pigmaeus?????
AGUCGCGUCGAAGCAGA--CGCAUGACGGAC
Homo_sapiens???????
CACAUUUU-CCUUGCAAAG
Pan_paniscus???????
CACAUCAU-CCUUGCAAAG
Gorilla_gorilla???? -ACAUCAUCCCUCGCAGAG
Pongo_pigmaeus?????
CACAUCAUCCCUUGCAGAG
;
endblock;
begin assumptions;
options deftype=unord;
?
9) Parsimony Jackknifer
The program by Farris is a parsimony program that also implements the
jackknife method to test the reliability of branches.
The format is similar to the MEGA format. On the first line a title/description
is placed in between single quotes. The alignment can be written in sequential
or interleaved format, but the sequence names have to be placed between
brackets. Also no blanks are allowed in the names. They should be replaced
by underscores ( _ ). The file is ended by a semicolon.
Examples:
' Four Anthropoidea '
(Homo_sapiens)???????
AGUCGAGUC---GCAGAAACGCAUGAC-GAC
CACAUUUU-CCUUGCAAAG
(Pan_paniscus)???????
AGUCGCGUCG--GCAGAAACGCAUGACGGAC
CACAUCAU-CCUUGCAAAG
(Gorilla_gorilla)????
AGUCGCGUCG--GCAGAUACGCAUCACGGAC
-ACAUCAUCCCUCGCAGAG
(Pongo_pigmaeus)?????
AGUCGCGUCGAAGCAGA--CGCAUGACGGAC
CACAUCAUCCCUUGCAGAG
;
---
' Four Anthropoidea '
(Homo_sapiens)???????
AGUCGAGUC---GCAGAAACGCAUGAC-GAC
(Pan_paniscus)???????
AGUCGCGUCG--GCAGAAACGCAUGACGGAC
(Gorilla_gorilla)????
AGUCGCGUCG--GCAGAUACGCAUCACGGAC
(Pongo_pigmaeus)?????
AGUCGCGUCGAAGCAGA--CGCAUGACGGAC
(Homo_sapiens)???????
CACAUUUU-CCUUGCAAAG
(Pan_paniscus)???????
CACAUCAU-CCUUGCAAAG
(Gorilla_gorilla)????
-ACAUCAUCCCUCGCAGAG
(Pongo_pigmaeus)?????
CACAUCAUCCCUUGCAGAG
;
10) PHYLIP
The PHYLIP package is a tree construction package that implements parsimony,
distance and maximum likelihood.
The format is pretty straightforward : on the first line the number
of sequences and their length (in characters) is displayed. Then the alignment
is displayed in an interleaved or sequential format. The sequence names
are allowed to contain blanks, but may not consist of more than 10 characters.
The interleaved format is slightly different from the other formats in
the way that the sequence names are only displayed in the first block,
while other interleaved formats repeat the names every block.
For example:
4 50
Homo sapie AGUCGAGUC---GCAGAAACGCAUGAC-GACC
Pan panisc AGUCGCGUCG--GCAGAAACGCAUGACGGACC
Gorilla go AGUCGCGUCG--GCAGAUACGCAUCACGGAC-
Pongo pigm AGUCGCGUCGAAGCAGA--CGCAUGACGGACC
ACAUUUU-CCUUGCAAAG
ACAUCAU-CCUUGCAAAG
ACAUCAUCCCUCGCAGAG
ACAUCAUCCCUUGCAGAG
The sequential format looks like this:
4 50
Homo sapie AGUCGAGUC---GCAGAAACGCAUGAC-GACC
ACAUUUU-CCUUGCAAAG
Pan panisc AGUCGCGUCG--GCAGAAACGCAUGACGGACC
ACAUCAU-CCUUGCAAAG
Gorilla go AGUCGCGUCG--GCAGAUACGCAUCACGGAC-
ACAUCAUCCCUCGCAGAG
Pongo pigm AGUCGCGUCGAAGCAGA--CGCAUGACGGACC
ACAUCAUCCCUUGCAGAG
You can find more info in the PHYLIP
package documentation.
11) TREECON
TREECON is a software package for construction and drawing of phylogenetic
trees on the basis of
evolutionary distances.
A full description of the TREECON format can be found right here.
Example:
50
Homo sapiens
AGUCGAGUC---GCAGAAACGCAUGAC-GACCACAUUUU-CCUUGCAAAG
Pan paniscus
AGUCGCGUCG--GCAGAAACGCAUGACGGACCACAUCAU-CCUUGCAAAG
Gorilla gorilla
AGUCGCGUCG--GCAGAUACGCAUCACGGAC-ACAUCAUCCCUCGCAGAG
Pongo pigmaeus
AGUCGCGUCGAAGCAGA--CGCAUGACGGACCACAUCAUCCCUUGCAGAG
?
?
?
Walk-through
?
?After succesfully installing ForCon, run the forcon.exe executable
(or just double-click the shortcut).
The start-up screen appears:

Pressing the 'Enter' button will continue the program.
First, you will be asked to specify the format of the input- and output
file:
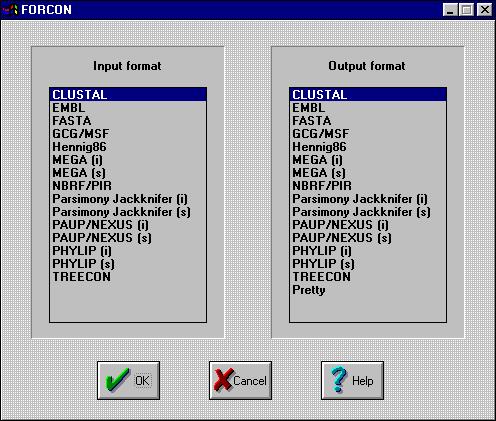
?
Just select the format from each list and press OK.
After doing this, you will be prompted to specify the input file.
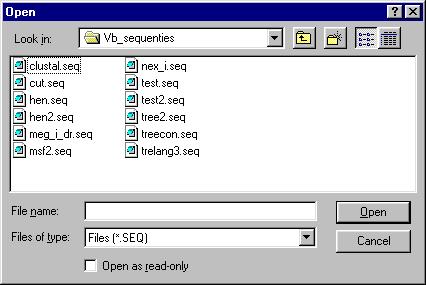
?
After this, the program will ask you for the blocksize/cutoff. Here
you can specify the number of characters that each block/sequence line
will consist of.
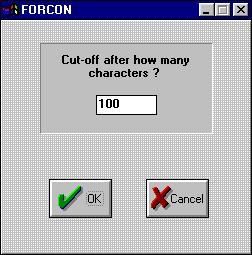
Fill in the text box and press OK.
If? your input file was a Hennig86 file, you are asked for the
'translation':
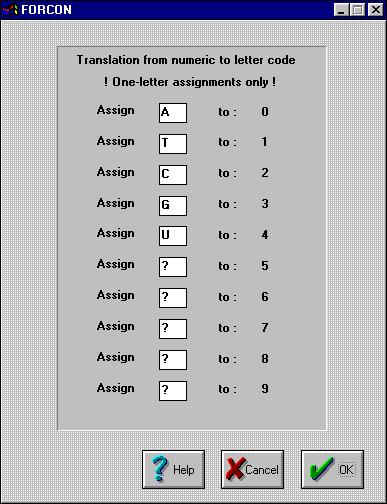
?
So, in this case, every 0 is translated into an A, 1 to T, etc.
Make sure just to enter one character for each box !
Specify the file you would like to save the new alignment in:
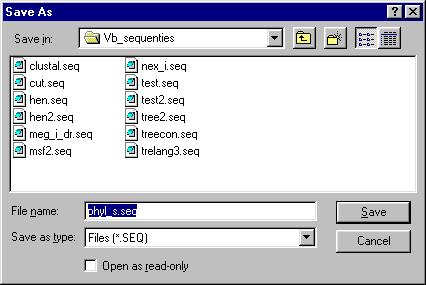
After doing this, you can make a selection of the sequences you would
like convert.
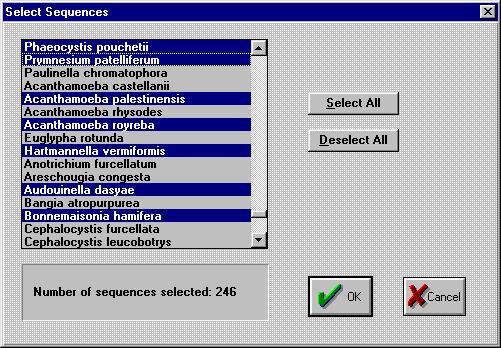
Click a name on the list to select that sequence. To select multiple
sequences, hold down the Control key on your keyboard while selecting.
Large blocks of sequences can be selected using the Shift key. Use the
Select All button to select all the sequences at once. The deselect all
button does the opposite.
After you made your selection, press OK.
You now get the chance to select certain positions of the alignment:
?
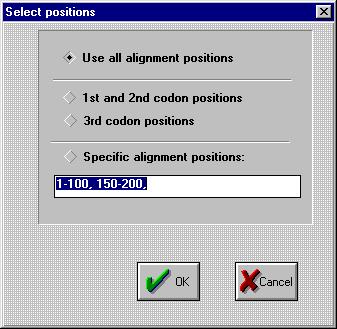
You can choose between 4 options:
?
-
use all of the alignment ( no change )
-
use the 1st and 2nd codon positions, e.g. AAU
GCU ACU ACG? becomes? AAGCACAC
-
only use the third codon positions, e.g. AAU
GCU ACU ACG? becomes UUUG
-
use specific user-defined codon positions: to cut parts out of your alignment;
areas should be separated by commas.
Just check the button of you choice, press OK, and we're off to:
?
The end.
You can find your file in the directory you specified earlier.
?
Disclaimer
This software is distributed freely 'as-is'. The programmer cannot be
held responsible for any damage that may occur. You can distribute the
program among your friends, colleagues, etc. in the original .ZIP file.
Please always register your program, if you should get a copy. It's free,
won't take much of your time, and you will be notified of any new releases
or bugs.
Yes, please,
register me !
If you encounter any bugs, please report them to me : jerae@gengenp.rug.ac.be
?
Acknowledgements
The programmers would like to thank ( in random order ) :
Julie Thompson, for her help on the CLUSTAL format
Rob Verschraegen, for his programming tips
Yves Van de Peer, for all his help
Alex Dong Li, for his help on the GCG/MSF format
All others who helped me in any way
?
?
Jeroen Raes
Research group of Bioinformatics
Department of Plant Genetics?? Tel:32.9.264 87 20??
Fax:32.9.264 50 08
University of Ghent, K.L. Ledeganckstraat 35, B-9000 GENT, Belgium
Laboratoire Associe de l'INRA
Vlaams interuniversitair Instituut voor Biotechnologie (VIB)
jerae@gengenp.rug.ac.be
to the ForCon
homepage
