Gene families were constructed as follows: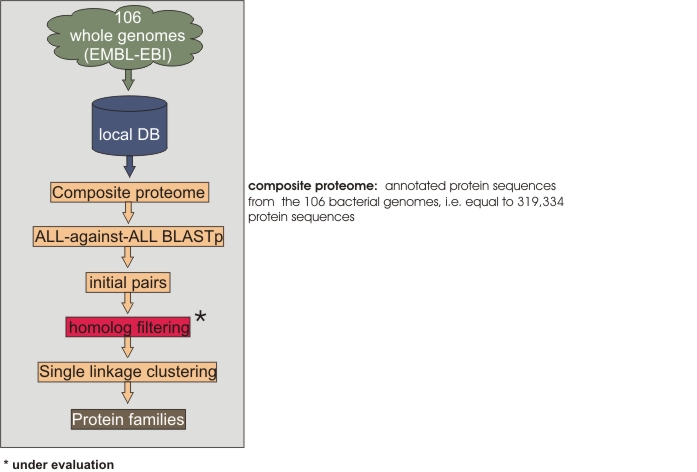
Homolog filtering was done on either of these 3 methods:
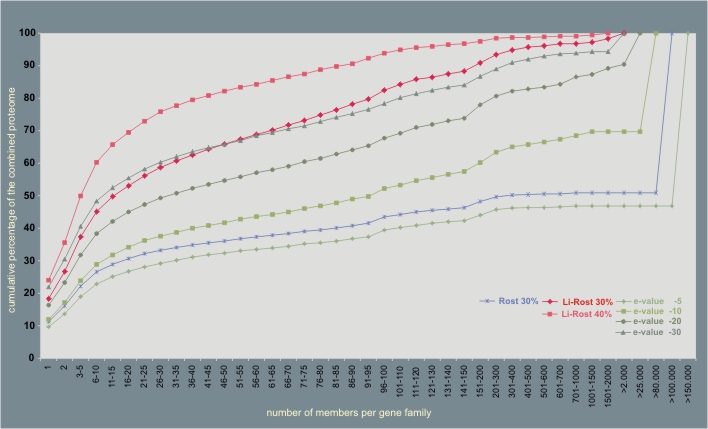
The effect of these different methods (with their different parameters) on the distribution of the proteome over a gene family size range is shown in the graph below. Stringent conditions (i.e. Li-Rost criterion) were found to be necessary in order to construct reliable gene families across genomes, as an e-value of 1e-10 for example resulted in one gene family of more than 80,000 members, i.e. 30% of our data set. We have chosen for the Li-Rost criterion (with the parameters set to > 30% identical residues) based on this distribution graph and an additional manual control for a number of specific gene families.In the combined bacterial proteome,excluding phage- and transposon-related ORFs, a total of 27,914 gene families were found, encompassing 243,910 proteins (81%).
References:Li, W.H. et al. (2001) Evolutionary analyses of the human genome. Nature 409, 847-849 Rost, B. (1999) Twilight zone of protein sequence alignments. Protein Eng 12, 85-94 |
|
|
Contact:
VIB / UGent Bioinformatics & Evolutionary Genomics Technologiepark 927 B-9052 Gent BELGIUM +32 (0) 9 33 13807 (phone) +32 (0) 9 33 13809 (fax) |