TREECON for Windows user
manual
DISTANCE ESTIMATION FOR NUCLEIC
ACID SEQUENCES
General
introduction and Jukes and Cantor model
Distance methods fit a tree to a matrix of N · (N-1)/2 pairwise
evolutionary distances, N being the number of sequences considered.
For every two sequences, the distance is a single value estimated from
the dissimilarity, i.e. the fraction of positions in which both sequences
differ. Due to the fact that some of these sequence differences are
the result of multiple events, this dissimilarity is actually an underestimation
of the true evolutionary distance. Therefore, one usually tries to
estimate the number of substitutions that have actually taken place by
applying a specific evolutionary model that makes assumptions about the
nature of evolutionary changes. However, since one does not have
an exact historical record of events that took place in the evolution of
sequences, correct estimation of the evolutionary distance is not self-evident.
One of the first substitution models used in the estimation of evolutionary
distances is the one of Jukes and Cantor (1969). This model
starts from the assumptions that all substitutions are independent, that
all sequence positions are equally subject to change, that substitutions
occur randomly among the four types of nucleotides, and that no insertions
or deletions have occurred. Based on these pre-assumptions, the authors
derived an equation for estimating evolutionary distances from observed
dissimilarity:
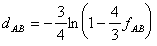 ,
,
where fAB is the dissimilarity (fraction of observed
differences) between sequences A and B, and dAB is the estimated
evolutionary distance (fraction of expected substitutions) between
sequences A and B. The relationship between dissimilarity and evolutionary
distance according to the Jukes and Cantor substitution model is shown
graphically in Fig. 1.
Several other equations for the estimation of evolutionary distances
have been proposed. For example, Kimura (1980) has provided a method
for inferring evolutionary distances based on a model of evolution
in which transitions and transversions may occur at different rates.
Other equations are based on substitution models in which the four different
nucleotides are not used in equal proportions (e.g. Tajima and Nei, 1984),
or where a bias in the direction of change is accounted for (e.g. Tamura
and Nei, 1993; Zharkikh, 1994).
An important drawback of most of these models is that they do not consider
differences in substitution rate among the sites of a molecule (see further).
The substitution model of Jukes and Cantor, also called the one-parameter
model, is the simplest one available for estimating the number of nucleotide
substitutions per site and is probably still the most used one.
When the fraction of differences between two
sequences exceeds 0.75, the distance cannot be computed. If f > 0.75,
TREECON crashes and gives a log-domain error. When this happens,
check your alignment and/or use the more conserved parts of the alignment
to construct a tree with.
Kimura -
two parameter model
Kimura (1980) provided a method for inferring
evolutionary distance in which transitions and transversions are treated
separately:
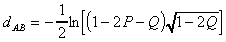
where P is the fraction of sequence positions
differing by a transition and Q is the fraction of sequence positions differing
by a transversion.
Tajima
& Nei
In the general correction of Tajima and Nei (1984),
the evolutionary distance is estimated by:

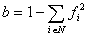
and fi is the frequency of the i-th
type of nucleotide belonging to the set of possible nucleotide types N
(= A, G, C, U or T) in the sequences being compared. This equation
holds for the model of nucleotide substitutions with equal substitution
rates between different nucleotides and does NOT take into account unequal
rates of substitution among different nucleotide pairs (Tajima and Nei,
1984). In TREECON, the computed base composition is the average for
all the sequences analyzed (as suggested in Swofford et al., 1996).
If the frequencies are 0.25 for all four nucleotides, this equation equals
the one of Jukes and Cantor.
Gamma
distances
All the previous distance measures start from the assumption that the
rate of nucleotide substitution is the same for all nucleotide sites.
However, in real sequences, this assumption rarely holds (see further).
Different studies suggest that the rate of nucleotide substitution varies
approximately according to the gamma distribution (see Uzzell and Corbin,
1971; Jin and Nei, 1990; Nei, 1991). This gamma distribution is specified
by a parameter a which is the square of the inverse of the coefficient
of variation of substitution rate (Nei, 1991).
For the Jukes and Cantor's one parameter model, the distance is computed
as follows (Jin and Nei, 1990):
for a=1:
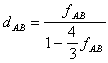
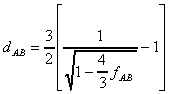
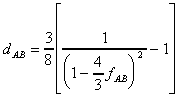
A more general equation has been given by
Rzhetsky and Nei (1994):

For the Kimura's two parameter evolution model, the distance is
computed as follows (Jin and Nei, 1990), where P is the fraction of
sequence positions differing by a transition and Q is the fraction of sequence
positions differing by a transversion:
for a=1:
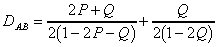
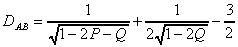
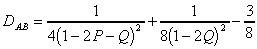
A more general equation has been given by
Nei (1991):
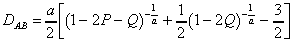
Galtier
& Gouy
The algorithm of Galtier and Gouy (1995) was developed for estimating
evolutionary distances without assuming homogeneity or stationarity of
the evolutionary process. This distance estimate should be useful
for phylogenetic analyses when compositional biases are observed in the
data. Two factors are taken into account: the transition/transversion
ratio, and the G+C content.
The transition/transversion ratio is assumed to be the same in all lineages.
It is estimated once, from the whole dataset. In the substitution
model of Galtier and Gouy, the ratio between the sums of transition and
of transversion rates equals a/2. This ratio is estimated for each
sequence pair (A, B):
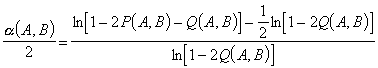
where P(A,B) is the observed proportion of sites in sequences A and
B showing a transition difference, and Q(A,B) is the observed proportion
of sites showing a transversion difference.
The estimate of parameter a is given by the mean of a(A,B) values for
all sequence pairs. This estimate is used for all pairwise distance
computations. In TREECON the transition/transversion ratio is estimated
before the actual computation of the evolutionary distances starts.
Since this value is based on all pairwise comparisons, this in fact doubles
the time needed for the estimation of distances between sequences.
In the bootstrap analysis, the transition/transversion ratio is only once
computed, based on the actual set of sequences.
The evolutionary distance is estimated as follows:
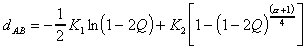
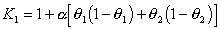 ,
,
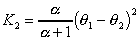
where theta1 Is the G+C content of sequence 1, and theta2 is the G+C
content of sequence 2.
Transversion
analysis
Sometimes, it can be interesting to estimate the evolutionary distance
on the basis of transversions only (see e.g. Woese et al., 1991; Van de
Peer et al., 1996b). The evolutionary distance is then estimated
by (Tajima and Nei, 1984; Swofford et al., 1996):
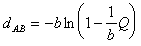
where Q is the fraction of transversions and
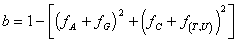
and fA + fG being the fraction of purines,
and fC + fU or fT being the fraction of
pyrimidines, computed over the complete alignment.
No correction
It is also possible not to correct for superimposed mutations (the
so-called p-distance)
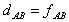
DISTANCE ESTIMATION FOR AMINO
ACID SEQUENCES
Overall, the computation of evolutionary distances for amino acid sequences
is similar to the computation of distances for nucleic acid sequences.
Poisson correction
The distance between two amino acid sequences is computed starting
from the assumption that the rate of amino acid substitution at each site
follows the poisson distribution (e.g. Zuckerkandl and Pauling, 1965; Dickerson,
1971):
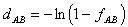
where fAB is the fraction of different amino acids between
two sequences (dissimilarity).
Kimura
This formula (Kimura, 1983) should be a good approximation to the Dayhoff
model (Hasegawa and Fujiwara, 1993):
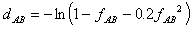
Tajima
& Nei
The evolutionary distance is calculated as:
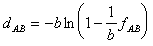
where b=0.95 (i.e. 19/20) for amino acid replacements (Tajima and Nei,
1984).
Gamma distances
When one starts from the assumption that the rate of amino acid substitutions
varies from site to site and follows the gamma distribution, the evolutionary
distance is computed as (Kumar et al., 1993; Ota and Nei, 1994):
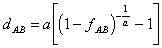
GENETIC DISTANCE ESTIMATION FOR
PATTERN ANALYSIS
At the moment, two methods to compute the genetic distance for pattern
analyses such as RFLP, RAPD, and AFLP are implemented.
Method 1 (Nei
and Li, 1979)
The genetic distance is computed as follows:
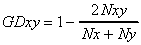 ,
,
where Nxy is the number of fragments (bands) shared in lines x and
y, and Nx is the number of fragments in line x, and Ny is the number of
fragments in line y.
Example
sample 1 1010100011
sample 1 1110011000
sample 2 1010111100
sample 2 1110000001
Nx=5
Nx=5
Ny=6
Ny=4
Nxy=3 Nxy=3
GDxy=1-(2*3)/(5+6)=0455
GDyx=1-(2*3)/(5+4)=0.33
Method
2 (Link et al., 1995)
The genetic distance is computed as follows:
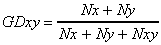 ,
,
where Nx is the number of bands in line x and not in line y, Ny
is the number of bands in line y and not in line x, and Nxy is the number
of bands shared in lines x and y.
Example
sample 1 1010100011
sample 1 1110011000
sample 2 1010111100
sample 2 1110000001
Nx=2
Nx=2
Ny=3
Ny=1
Nxy=3
Nxy=3
GDxy=(2+3)/(2+3+3)=5/8=0.625
GDyx=(2+1)/(2+1+3)=3/6=0.5
INSERTIONS
AND DELETIONS
In TREECON, the user can choose whether to take insertions and deletions
(indels) into account or not. If they are, they are counted separately
since the correction for superimposed events does not hold for insertions
and deletions. For example, in TREECON, the evolutionary distance
according to the Jukes and Cantor model is then computed as (Van de Peer
et al., 1990):
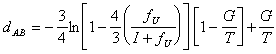
where I is the number of identical nucleotides, fU is the
number of positions showing a substitution and G is the number of gaps
in one sequence with respect to the other. T is the sum of I, fU
and G. The first term of the equation accounts for substitutions
and comprises the Jukes and Cantor correction factor for multiple mutations
per site (see above). The second term accounts for deletions and
insertions. A row of adjacent gaps is treated one gap, regardless
of its length. Ambiguities (not A, G, C, T or U) are not taken into
account. Comparison of the imaginary sequences:
A G C U G G - - - G C N U A
- - C U A G A A A G A C U A
would give the values I=6, fU=2, G=2, and T=10.
HOW TO
CHECK DISTANCE ESTIMATES
Distance estimates are saved in the file mat.out. The format
of saving may seem a bit awkward but this is for compatibility reasons
with other (older) software.
If one wants to know the evolutionary distance between two sequences,
this is how to interpret the distance matrix (5 sequences were compared):
Tree construction
(title)
5 0
0 2 0 (not
important, except first digit)
(5f8.3)
(not important)
19.534 0.000
26.837 25.016 24.728 (sequence 2)
24.304 26.837
0.000 9.079 20.176 (sequence 3)
24.593 25.016
9.079 0.000 19.053 (sequence 4)
25.280 24.728 20.176
19.053 0.000 (sequence 5)
(seq 1) (seq 2) (seq 3) (seq
4) (seq 5)
For example, the evolutionary distance (number of substitutions per
nucleotide) between sequences 2 and 4 equals 0.25016 (the values in the
distance matrix are multiplied by 100).
In future, I will make it possible to display the distance estimates
in a more user-friendly way.
Return