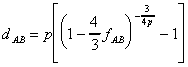 ,
,SUBSTITUTION RATE CALIBRATION
However, differences in substitution rate among sites is very common in almost all genes. In the case of ribosomal RNA, the substitution rate was estimated (Van de Peer et al. 1993b, 1996a, c) to vary by a factor of more than 1000 from the least variable to the most variable site. Olsen (1987) already demonstrated that application of the Jukes and Cantor correction to sequences composed of sites with unequal rates can lead to artefacts in tree topology, caused by the underestimation of the distances, and proposed a different evolutionary model that assumed a log-normal distribution of substitution rates over the sequence positions. Jin and Nei (1990) followed a similar approach but assumed that substitution rates were gamma distributed (see above). On the basis of this gamma distribution, which involves a parameter a that describes the extent of the rate variation, they derived several equations to compute the evolutionary distance from the observed sequence dissimilarities. However, the problem with applying gamma distances is the estimation of the gamma parameter a, the accuracy of which depends on the estimation method, tree topology, and on the number of sequences used in the estimation (Rzhetsky and Nei, 1994; Yang, 1996; Sullivan et al., 1996; Tourasse and Gouy, 1997).
One of the main problems in considering among-site rate variation is the quantitative estimation of the substitution rates or variabilities of nucleotide sites. Maximum parsimony estimates can be heavily biased (Wakeley, 1993; Yang, 1996; Tourasse and Gouy, 1997) while maximum likelihood estimates may experience computational difficulties (Yang, 1996). Recently, we developed a method for measuring the relative substitution rate of individual sites in a nucleotide sequence alignment on the basis of a distance approach (Van de Peer et al., 1993b; Van de Peer et al., 1996c). The main advantage of this approach is that nucleotide variability estimates do not depend upon a given tree topology, contrary to estimates inferred from maximum parsimony or maximum likelihood methods (Sullivan et al., 1996; Yang, 1996), and that they can be based on an very large number of sequences. This is important since the more sequences taken into account, the more accurate the substitution rates can be estimated (see also Tourasse and Gouy, 1997). When the substitution rates of the individual nucleotides of the molecule are computed as described previously (Van de Peer et al., 1993b, 1996c), an equation can be derived that better describes the actual relationship between sequence dissimilarity and evolutionary distance:
,Substitution rate calibration has shown to be particularly useful in eliminating artificial clustering caused by increased evolutionary rates. In many cases, higher evolutionary rates tend to pull sequences closer to the base of the tree. Moreover, due to the serious underestimation of large evolutionary distances (see figure 2), distant species seem closer to one another than they actually are, which often result in artificial clustering of long branches, the so called long branch attraction phenomenon.
How does substitution rate calibration works?
For an alignment of N sequences, N · (N-1)/2 pairwise evolutionary distances d are computed according to the equation of Jukes and Cantor (see above).
When all pairwise distances are computed, they are classified into a number of distance intervals, e.g. distances from 0 to 0.005, distances from 0.005 to 0.010, and so on. Next, the fraction of sequence pairs falling within a certain distance interval and characterized by a nucleotide change, is computed. This is done for every nucleotide position. This fraction is then plotted against the mean of the distance interval (Van de Peer et al., 1993). A curve obeying equation
Actually, the estimated nucleotide substitution rates are not yet optimal, because they are derived on the basis of a distance matrix computed by means of equation of Jukes and Cantor. This equation only gives a first approximation of the relation between dissimilarity and distance, since it starts from the unrealistic assumption that all nucleotides have the same substitution rate. Therefore, after estimation of all vi values, alignment positions are grouped into sets of similar variability. A spectrum of relative nucleotide substitution rates is thus obtained (see Fig. 4). Once the shape of the spectrum is known, it is possible to derive the following equation for the dissimilarity, f, as a function of the evolutionary distance d (Van de Peer et al., 1996 (see appendix)):
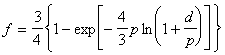
The value of parameter p depends on the shape of the substitution rate spectrum. The inverse of this equation,
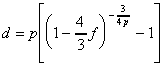 ,
,This iterative process is repeated several times until the nucleotide substitution rates vi do not change anymore (see Fig. 5). In general, no more than 3 iterations are needed for the changes to become imperceptible.
A practical example on rate calibration
Heres how to proceed:
In the substitution rate calibration method, a new function to convert
dissimilarity into distance is derived considering only positions for which
a variability can be estimated. Absolutely conserved positions (for
which the variability = 0) are thus not considered. Therefore, by default,
these positions are also left out of consideration during the actual computation
of evolutionary distances. Positions that are deleted in > 50% of
the sequences are not considered either.
Now compare this tree with a tree constructed by using the Jukes and Cantor (or Kimura) correction on the basis of the file TEST_CAL.SEQ. As you can see, the position of the fastly evolving sequences (long branches) is quite different in both trees.
Files that may contain valuable information are:
Substitution rate calibration should only be applied when sufficient sequences are available, and of sufficient length. Rate calibration based on 20 sequences of about 500 nucleotides is not very reliable. Use at least 35-40 sequences, and with > 1000 positions that are variable. If you want to construct a tree of only 10 sequences with substitution rate calibration, try to estimate nucleotide variabilities on a larger set of sequences. Once the rate spectrum is inferred and the function derived (parameter "p" has been computed), these can be used on a subset of sequences.
Of course, substitution rate calibration is not a miracle method that solves all artifacts caused by fastly evolving species. However, in several cases it does give an improved tree topology. When clear differences are observed between trees constructed with rate calibration and trees constructed by standard corrections for multiple mutations, increased evolutionary rates in certain lineages are probably at play.
Applications of the substitution rate calibration method can be found
in: