
Research goal
The decreasing cost of genome sequencing enables charting the genomes of large cohorts of individuals. Integrating molecular features obtained from cohorts of individuals with corresponding phenotypic traits offers vast potential to understand the molecular mechanisms affecting complex traits, but also to study their evolution. Our research focuses on developing integrative methods that can exploit natural genomic variation to better understand adaptive traits and their evolution. Key to our methods is the combination of domain knowledge with advanced statistics and datamining. As methods have to be useful for users (biologists, clinicians) we focus on interpretable models.
Research topics on Bioinformatics method development
Network Inference
Publicly available omics data provide a useful resource to infer molecular interaction networks, in which nodes represent genes and edges the interactions between the genes. We have done pioneering work in the domain of motif detection, coexpression analysis and data integration to infer interaction networks.
Example publications:
Network-based data interpretation
 Overlaying such interaction networks with in house generated data provide a comprehensive and intuitive way to inteprete data. In our research we are developing graph-based datamining procedures that can use these networks as a scaffold for in house data interpretation.
Overlaying such interaction networks with in house generated data provide a comprehensive and intuitive way to inteprete data. In our research we are developing graph-based datamining procedures that can use these networks as a scaffold for in house data interpretation.
Example publication:
Systems Genetics: identifying pathways associating with adaptive traits
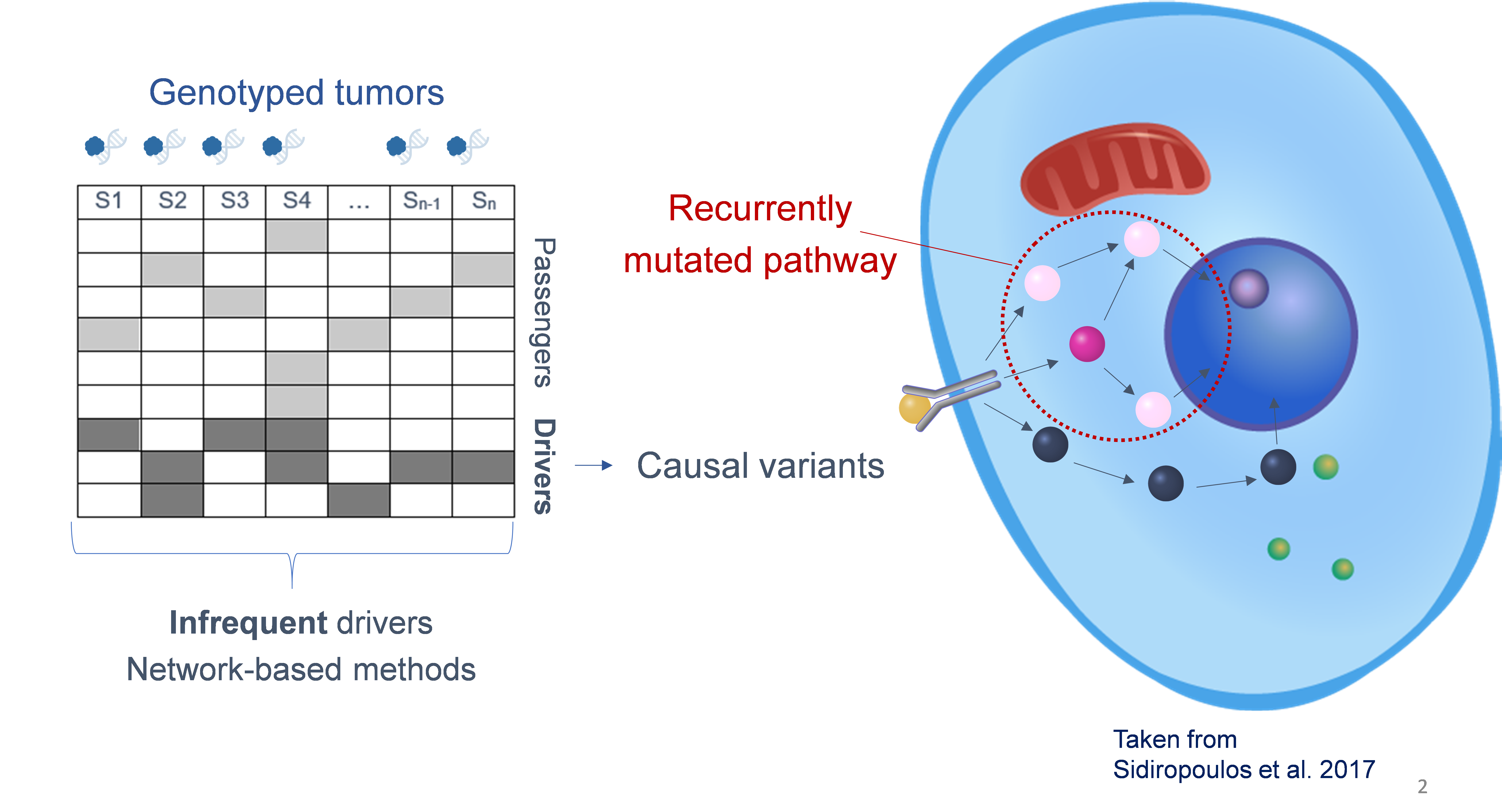 We develop alternative techniques that incorporate biological domain knowledge in the form of interaction networks to facilitate genotype-phenotype mapping. These methods use graph-based and deep learning techniques to search for genes carrying variants that are tightly ‘connected’ to each other and to the genes involved in a downstream expression phenotype on an interaction network.
We develop alternative techniques that incorporate biological domain knowledge in the form of interaction networks to facilitate genotype-phenotype mapping. These methods use graph-based and deep learning techniques to search for genes carrying variants that are tightly ‘connected’ to each other and to the genes involved in a downstream expression phenotype on an interaction network.
Evolutionary reconstruction of clonal systems
 Clonal population (cancer, bacterial populations) are heterogeneous. During evolution adaptive mutations originate that drive selection sweeps. High coverage population based sequencing of such heterogeneous communities allows reconstructing the individual subpopulations and tracing their evolution. In our research we are developing methods for reconstruction of clonal subpopulations, with a main focus on bacterial populations.
Clonal population (cancer, bacterial populations) are heterogeneous. During evolution adaptive mutations originate that drive selection sweeps. High coverage population based sequencing of such heterogeneous communities allows reconstructing the individual subpopulations and tracing their evolution. In our research we are developing methods for reconstruction of clonal subpopulations, with a main focus on bacterial populations.
Example publication: